

Bcl-2 and Bcl-XL serve an anti-inflammatory function in endothelial cells through inhibition of NF-kappaB
- PMID: 10021463
- PMCID: PMC408093
- DOI: 10.1172/JCI2517
Bcl-2 and Bcl-XL serve an anti-inflammatory function in endothelial cells through inhibition of NF-kappaB
Abstract
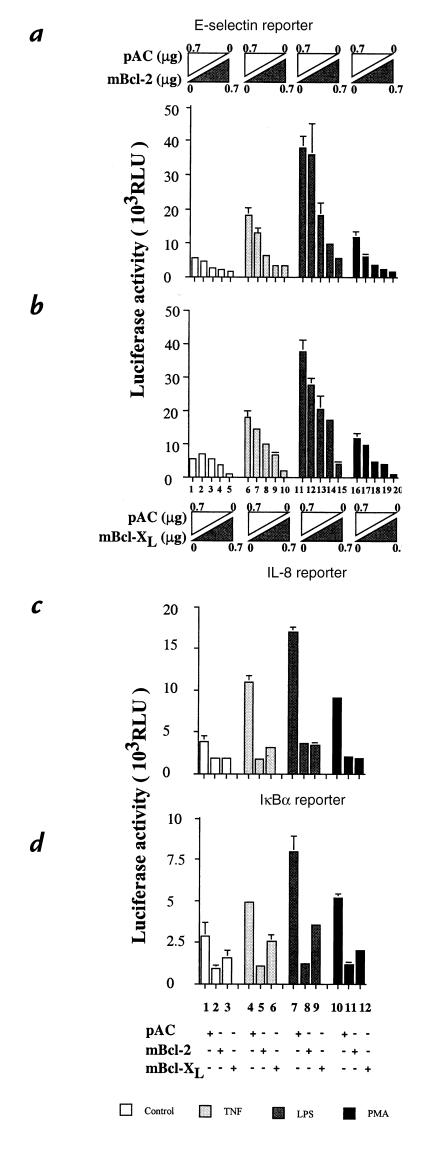
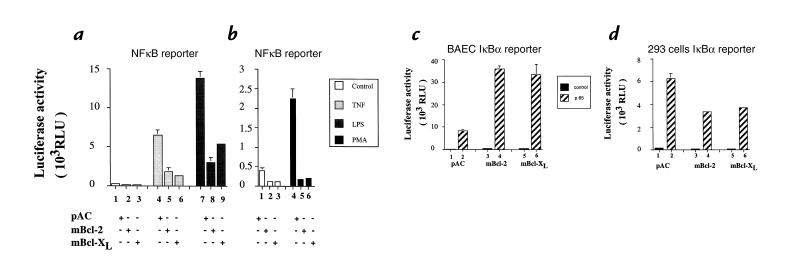
To maintain the integrity of the vascular barrier, endothelial cells (EC) are resistant to cell death. The molecular basis of this resistance may be explained by the function of antiapoptotic genes such as bcl family members. Overexpression of Bcl-2 or Bcl-XL protects EC from tumor necrosis factor (TNF)-mediated apoptosis. In addition, Bcl-2 or Bcl-XL inhibits activation of NF-kappaB and thus upregulation of proinflammatory genes. Bcl-2-mediated inhibition of NF-kappaB in EC occurs upstream of IkappaBalpha degradation without affecting p65-mediated transactivation. Overexpression of bcl genes in EC does not affect other transcription factors. Using deletion mutants of Bcl-2, the NF-kappaB inhibitory function of Bcl-2 was mapped to bcl homology domains BH2 and BH4, whereas all BH domains were required for the antiapoptotic function. These data suggest that Bcl-2 and Bcl-XL belong to a cytoprotective response that counteracts proapoptotic and proinflammatory insults and restores the physiological anti-inflammatory phenotype to the EC. By inhibiting NF-kappaB without sensitizing the cells (as with IkappaBalpha) to TNF-mediated apoptosis, Bcl-2 and Bcl-XL are prime candidates for genetic engineering of EC in pathological conditions where EC loss and unfettered activation are undesirable.
Figures




References
-
- Pober JS, et al. The potential roles of vascular endothelium in immune reactions [review] Hum Immunol. 1990;28:258–262. - PubMed
-
- Cotran RS, Pober JS. Cytokine-endothelial interactions in inflammation, immunity, and vascular injury. J Am Soc Nephrol. 1990;1:225–235. - PubMed
-
- Bach FH, et al. Endothelial cell activation and thromboregulation during xenograft rejection [review] Immunol Rev. 1994;141:5–30. - PubMed
-
- Gimbrone MAJ. Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis [review] Am J Cardiol. 1995;75:67B–70B. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
