

DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model
- PMID: 10021468
- PMCID: PMC408103
- DOI: 10.1172/JCI5346
DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model
Abstract
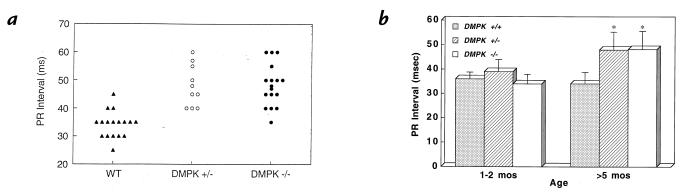
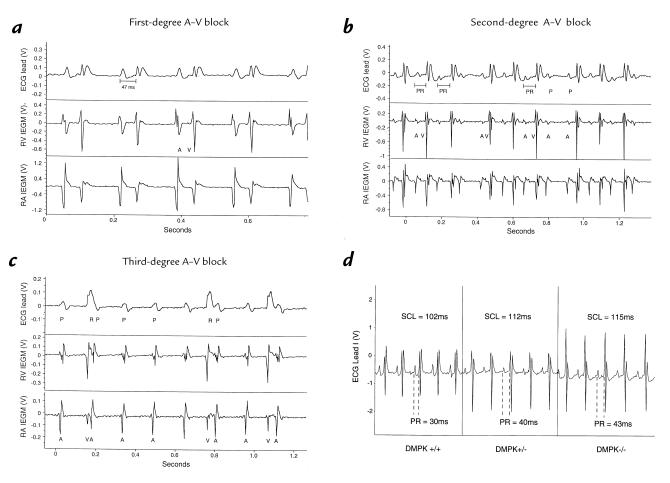
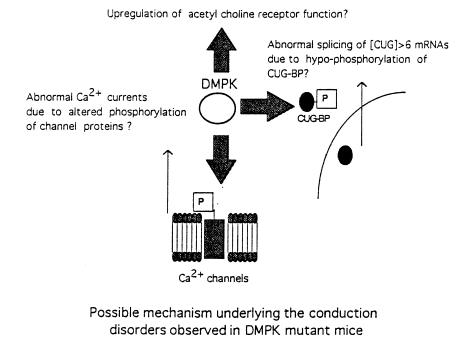
Myotonic dystrophy (DM) is the most common form of muscular dystrophy and is caused by expansion of a CTG trinucleotide repeat on human chromosome 19. Patients with DM develop atrioventricular conduction disturbances, the principal cardiac manifestation of this disease. The etiology of the pathophysiological changes observed in DM has yet to be resolved. Haploinsufficiency of myotonic dystrophy protein kinase (DMPK), DM locus-associated homeodomain protein (DMAHP) and/or titration of RNA-binding proteins by expanded CUG sequences have been hypothesized to underlie the multi-system defects observed in DM. Using an in vivo murine electrophysiology study, we show that cardiac conduction is exquisitely sensitive to DMPK gene dosage. DMPK-/- mice develop cardiac conduction defects which include first-, second-, and third-degree atrioventricular (A-V) block. Our results demonstrate that the A-V node and the His-Purkinje regions of the conduction system are specifically compromised by DMPK loss. Importantly, DMPK+/- mice develop first-degree heart block, a conduction defect strikingly similar to that observed in DM patients. These results demonstrate that DMPK dosage is a critical element modulating cardiac conduction integrity and conclusively link haploinsufficiency of DMPK with cardiac disease in myotonic dystrophy.
Figures
Similar articles
-
Progressive atrioventricular conduction block in a mouse myotonic dystrophy model.J Interv Card Electrophysiol. 2000 Jun;4(2):351-8. doi: 10.1023/a:1009842114968. J Interv Card Electrophysiol. 2000. PMID: 10936001
-
Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice.Proc Natl Acad Sci U S A. 1997 Nov 25;94(24):13221-6. doi: 10.1073/pnas.94.24.13221. Proc Natl Acad Sci U S A. 1997. PMID: 9371827 Free PMC article.
-
Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy.Nat Genet. 2006 Sep;38(9):1066-70. doi: 10.1038/ng1857. Epub 2006 Jul 30. Nat Genet. 2006. PMID: 16878132 Free PMC article.
-
Myotonic dystrophy and myotonic dystrophy protein kinase.Prog Histochem Cytochem. 2000;35(3):187-251. doi: 10.1016/s0079-6336(00)80002-9. Prog Histochem Cytochem. 2000. PMID: 11064921 Review.
-
[DM pathogenesis and expansion of CTG trinucleotide repeat within 3' untranslated region of DMPK gene].Nihon Rinsho. 1999 Apr;57(4):937-42. Nihon Rinsho. 1999. PMID: 10222792 Review. Japanese.
Cited by
-
Cardiac Pathology in Myotonic Dystrophy Type 1.Int J Mol Sci. 2021 Nov 2;22(21):11874. doi: 10.3390/ijms222111874. Int J Mol Sci. 2021. PMID: 34769305 Free PMC article. Review.
-
Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1.J Pharmacol Exp Ther. 2015 Nov;355(2):329-40. doi: 10.1124/jpet.115.226969. Epub 2015 Sep 1. J Pharmacol Exp Ther. 2015. PMID: 26330536 Free PMC article.
-
Epigenetics of the myotonic dystrophy-associated DMPK gene neighborhood.Epigenomics. 2016 Jan;8(1):13-31. doi: 10.2217/epi.15.104. Epub 2016 Jan 12. Epigenomics. 2016. PMID: 26756355 Free PMC article.
-
Progressive atrioventricular conduction block in a mouse myotonic dystrophy model.J Interv Card Electrophysiol. 2000 Jun;4(2):351-8. doi: 10.1023/a:1009842114968. J Interv Card Electrophysiol. 2000. PMID: 10936001
-
Myotonic dystrophy mouse models: towards rational therapy development.Trends Mol Med. 2011 Sep;17(9):506-17. doi: 10.1016/j.molmed.2011.05.004. Epub 2011 Jul 2. Trends Mol Med. 2011. PMID: 21724467 Free PMC article. Review.
References
-
- O’Brien TA, Harper PS. Course, prognosis and complications of childhood-onset myotonic dystrophy. Dev Med Child Neurol. 1984;26:62–67. - PubMed
-
- Harper, P.S. 1989. Myotonic Dystrophy. 2nd ed. W.B. Saunders. London, United Kingdom.
-
- Brook JD, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. - PubMed
-
- Fu Y-H, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. - PubMed
-
- Mahadevan M, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
