

A low-affinity serum response element allows other transcription factors to activate inducible gene expression in cardiac myocytes
- PMID: 10022871
- PMCID: PMC83977
- DOI: 10.1128/MCB.19.3.1841
A low-affinity serum response element allows other transcription factors to activate inducible gene expression in cardiac myocytes
Abstract
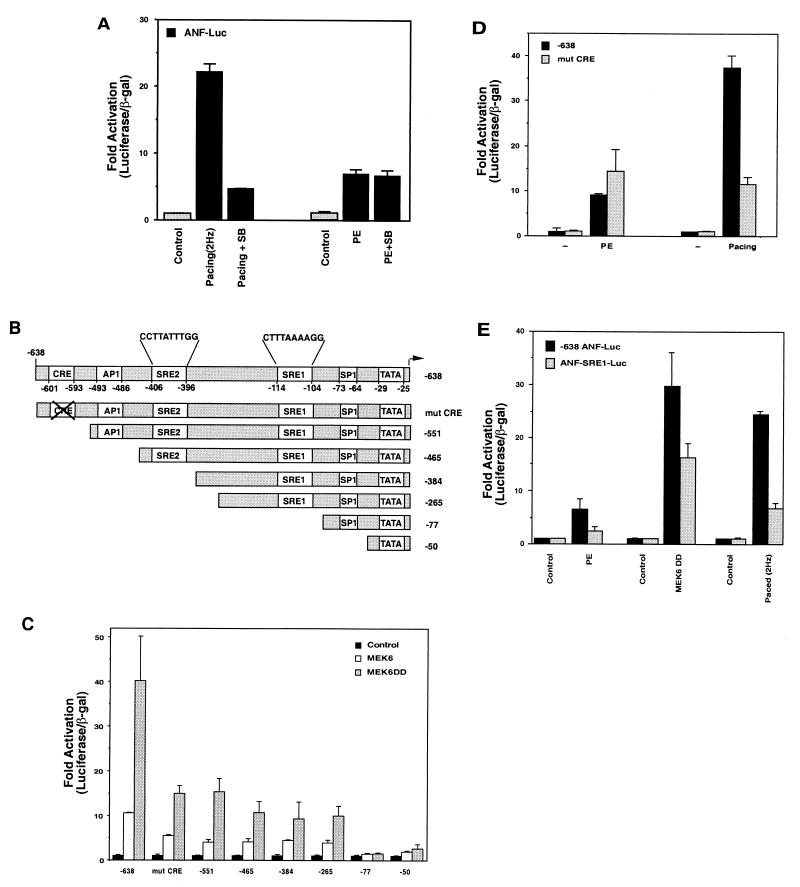
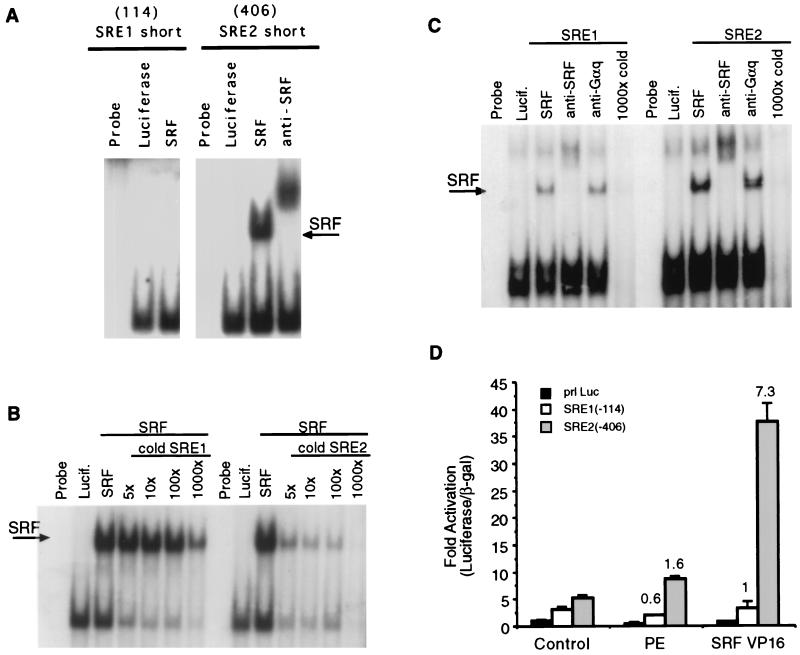
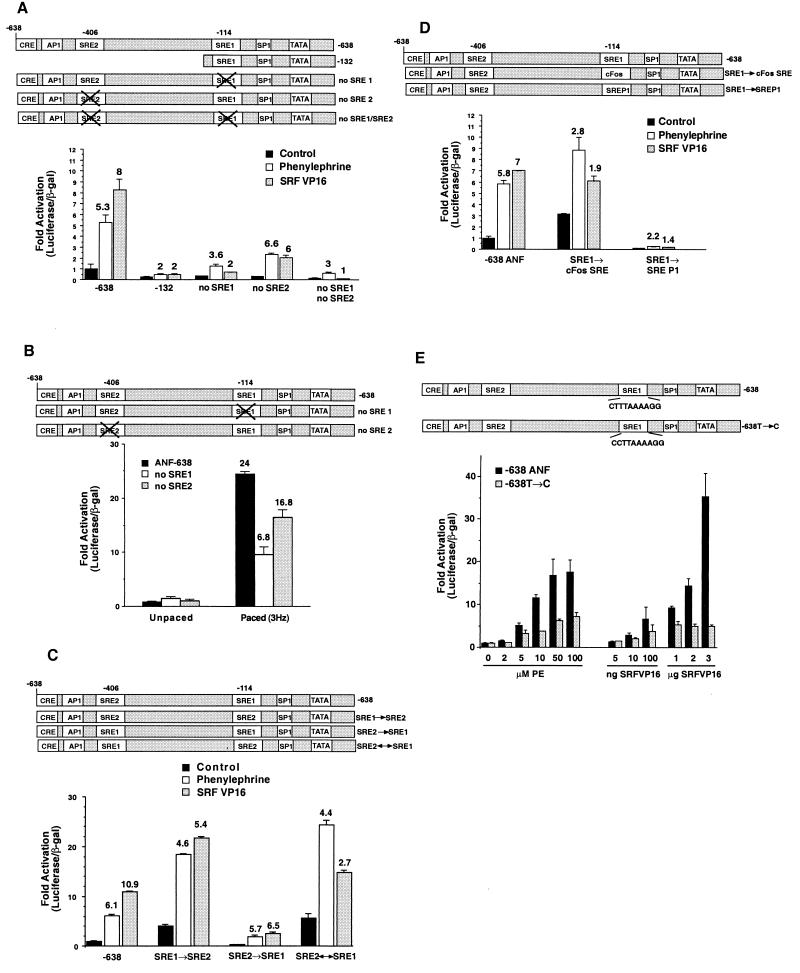
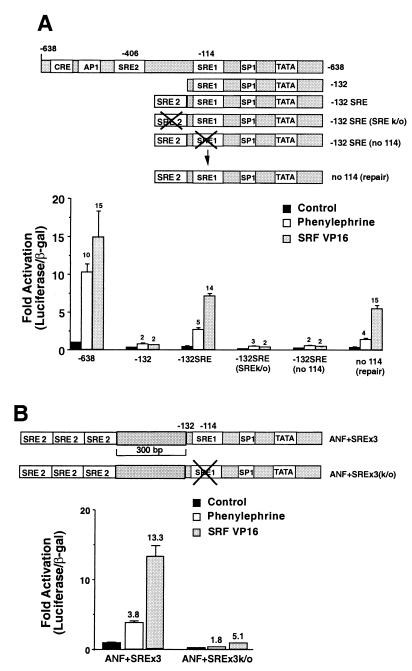
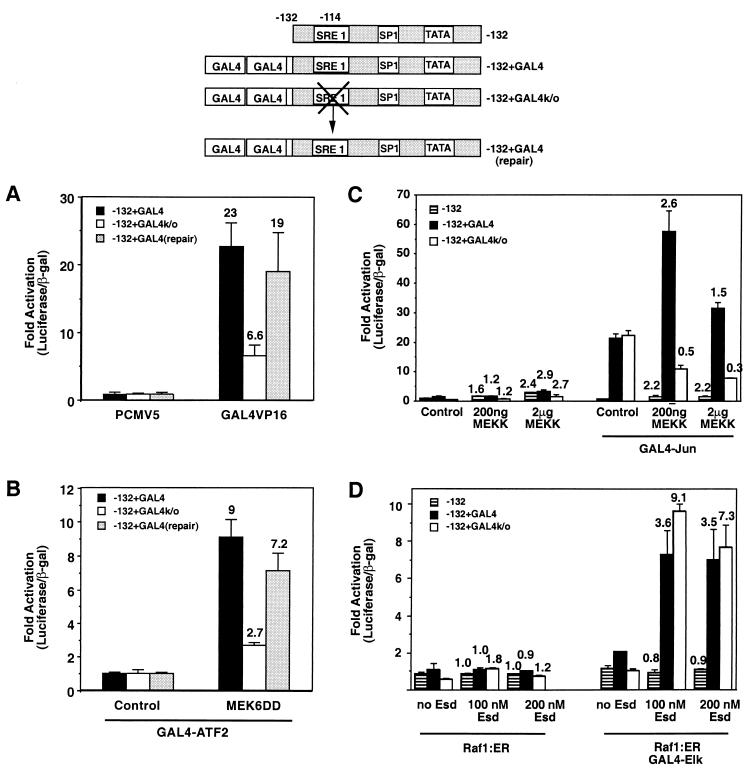
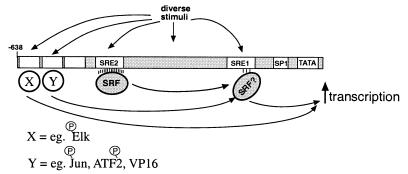
Hypertrophic growth of cardiac muscle cells is induced by a variety of physiological and pathological stimuli and is associated with a number of changes, including activation of genes such as atrial natriuretic factor. We found that two serum response element (SRE)-like DNA elements, one of which does not meet the consensus sequence and binds serum response factor (SRF) with low affinity, regulate the activity of this promoter. Surprisingly, the ability to induce the promoter by two different physiologic stimuli, as well as various activated transcription factors, including SRF-VP16, was primarily dependent upon the nonconsensus rather than the consensus SRE. This SRE controls the induction of gene expression via an unusual mechanism in that it is required to allow some, but not all, active transcription factors at unrelated sites on the promoter to stimulate gene expression. Thus, in addition to regulation of SRF activity by growth stimuli, regulation of a low-affinity SRE element controls inducible gene expression by modulating the ability of other transcription factors to stimulate the transcription machinery.
Figures
Similar articles
-
p38 Mitogen-activated protein kinase mediates the transcriptional induction of the atrial natriuretic factor gene through a serum response element. A potential role for the transcription factor ATF6.J Biol Chem. 1998 Aug 7;273(32):20636-43. doi: 10.1074/jbc.273.32.20636. J Biol Chem. 1998. PMID: 9685422
-
Transforming growth factor-beta1 and protein kinase C synergistically activate the c-fos serum response element in myocardial cells.J Mol Cell Cardiol. 1998 Mar;30(3):551-62. doi: 10.1006/jmcc.1997.0619. J Mol Cell Cardiol. 1998. PMID: 9515031
-
L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway.J Biol Chem. 1994 Oct 14;269(41):25483-93. J Biol Chem. 1994. PMID: 7929249
-
Growth factors, growth factor response elements, and the cardiac phenotype.Basic Res Cardiol. 1992;87 Suppl 2:33-48. doi: 10.1007/978-3-642-72477-0_4. Basic Res Cardiol. 1992. PMID: 1284369 Review.
-
Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression.J Cell Biochem. 2004 Sep 1;93(1):74-82. doi: 10.1002/jcb.20199. J Cell Biochem. 2004. PMID: 15352164 Review.
Cited by
-
The neuron-restrictive silencer element-neuron-restrictive silencer factor system regulates basal and endothelin 1-inducible atrial natriuretic peptide gene expression in ventricular myocytes.Mol Cell Biol. 2001 Mar;21(6):2085-97. doi: 10.1128/MCB.21.6.2085-2097.2001. Mol Cell Biol. 2001. PMID: 11238943 Free PMC article.
-
Target gene-specific modulation of myocardin activity by GATA transcription factors.Mol Cell Biol. 2004 Oct;24(19):8519-28. doi: 10.1128/MCB.24.19.8519-8528.2004. Mol Cell Biol. 2004. PMID: 15367672 Free PMC article.
-
Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin.Mol Cell Biol. 2005 Jan;25(1):364-76. doi: 10.1128/MCB.25.1.364-376.2005. Mol Cell Biol. 2005. PMID: 15601857 Free PMC article.
-
Myocardin expression is regulated by Nkx2.5, and its function is required for cardiomyogenesis.Mol Cell Biol. 2003 Dec;23(24):9222-32. doi: 10.1128/MCB.23.24.9222-9232.2003. Mol Cell Biol. 2003. PMID: 14645532 Free PMC article.
-
Degeneration of the mouse retina upon dysregulated activity of serum response factor.Mol Vis. 2011 Apr 29;17:1110-27. Mol Vis. 2011. PMID: 21552476 Free PMC article.
References
-
- Akhter S A, Luttrell L M, Rockman H A, Iaccarino G, Lefkowitz R J, Koch W J. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577. - PubMed
-
- Alberts A S, Geneste O, Treisman R. Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell. 1998;92:475–487. - PubMed
-
- Allo S N, McDermott P J, Carl L L, Morgan H E. Phorbol ester stimulation of protein kinase C activity and ribosomal DNA transcription: role in hypertrophic growth of cultured cardiomyocytes. J Biol Chem. 1991;266:22003–22009. - PubMed
-
- Bogoyevitch M A, Gillespie-Brown J, Ketterman A J, Fuller S J, Ben-Levy R, Ashworth A, Marshall C J, Sugden P H. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. Circ Res. 1996;79:162–173. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous