

Safety and pharmacokinetics of abacavir (1592U89) following oral administration of escalating single doses in human immunodeficiency virus type 1-infected adults
- PMID: 10049274
- PMCID: PMC89167
- DOI: 10.1128/AAC.43.3.603
Safety and pharmacokinetics of abacavir (1592U89) following oral administration of escalating single doses in human immunodeficiency virus type 1-infected adults
Abstract
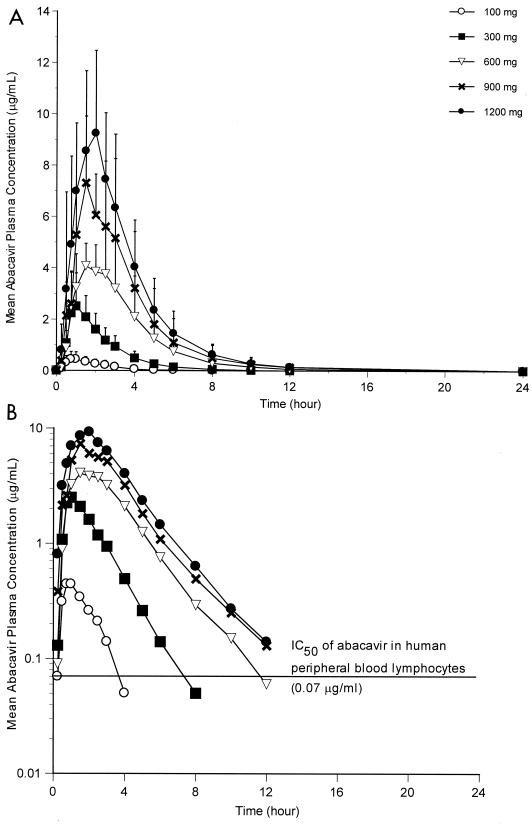
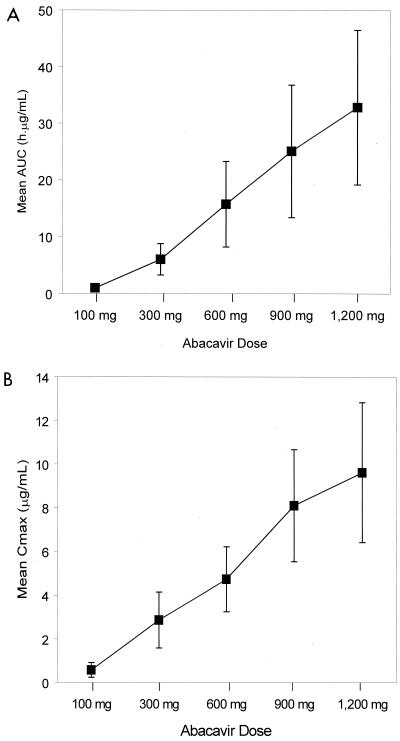
Abacavir (1592U89) is a nucleoside analog reverse transcriptase inhibitor that has been demonstrated to have selective activity against human immunodeficiency virus (HIV) in vitro and favorable safety profiles in mice and monkeys. A phase I study was conducted to evaluate the safety and pharmacokinetics of abacavir following oral administration of single escalating doses (100, 300, 600, 900, and 1,200 mg) to HIV-infected adults. In this double-blind, placebo-controlled study, subjects with baseline CD4+ cell counts ranging from < 50 to 713 cells per mm3 (median, 315 cells per mm3) were randomly assigned to receive abacavir (n = 12) or placebo (n = 6). The bioavailability of the caplet formulation relative to that of the oral solution was also assessed with the 300-mg dose. Abacavir was well tolerated by all subjects; mild to moderate asthenia, abdominal pain, headache, diarrhea, and dyspepsia were the most frequently reported adverse events, and these were not dose related. No significant clinical or laboratory abnormalities were observed throughout the study. All doses resulted in mean abacavir concentrations in plasma that exceeded the mean 50% inhibitory concentration (IC50) for clinical HIV isolates in vitro (0.07 microgram/ml) for almost 3 h. Abacavir was rapidly absorbed following oral administration, with the time to the peak concentration in plasma occurring at 1.0 to 1.7 h postdosing. Mean maximum concentrations in plasma (Cmax) and the area under the plasma concentration-time curve from time zero to infinity (AUC0-infinity) increased slightly more than proportionally from 100 to 600 mg (from 0.6 to 4.7 micrograms/ml for Cmax; from 1.0 to 15.7 micrograms.h/ml for AUC0-infinity) but increased proportionally from 600 to 1,200 mg (from 4.7 to 9.6 micrograms/ml for Cmax; from 15.7 to 32.8 micrograms.h/ml for AUC0-infinity. The elimination of abacavir from plasma was rapid, with an apparent elimination half-life of 0.9 to 1.7 h. Abacavir was well absorbed, with a relative bioavailability of the caplet formulation of 96% versus that of an oral solution (drug substance in water). In conclusion, this study showed that abacavir is safe and is well tolerated by HIV-infected subjects and demonstrated predictable pharmacokinetic characteristics when it was administered as single oral doses ranging from 100 to 1,200 mg.
Figures
References
-
- Child S, Montaner J, Tsoukas C, Fanning M, Le T, Wall R A, Ruedy J. Canadian multicenter azidothymidne trial: AZT pharmacokinetics. J Acquired Immune Defic Syndr. 1991;4:865–870. - PubMed
-
- Ching S V, Ayers K M, Dornsife R E, Grebe G L, Howard J L. Abstracts of the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy. Washington, D.C: American Society for Microbiology; 1994. Nonclinical toxicology and in vitro toxicity studies with the novel anti-HIV agent (1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol (1592U89) succinate, abstr. I88; p. 92.
-
- Daluge S M, Good S S, Faletto M B, Miller W H, St. Clair M H, Boone L R, Tisdale M, Parry N R, Reardon J E, Dornsife R E, Averett D R, Krenitsky T A. 1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity. Antimicrob Agents Chemother. 1997;41:1082–1093. - PMC - PubMed
-
- Dudley M N, Graham K K, Kaul S, Geletko S, Dunkle L, Browne M, Mayer K. Pharmacokinetics of stavudine in patients with AIDS or AIDS-related complex. J Infect Dis. 1992;166:480–485. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
