

Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage
- PMID: 10051617
- PMCID: PMC26759
- DOI: 10.1073/pnas.96.5.2192
Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage
Abstract
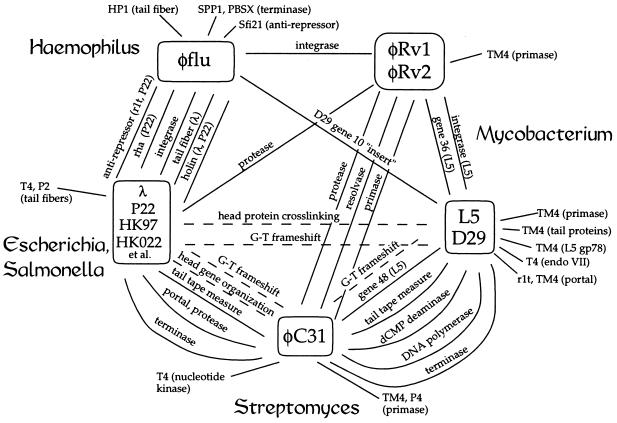
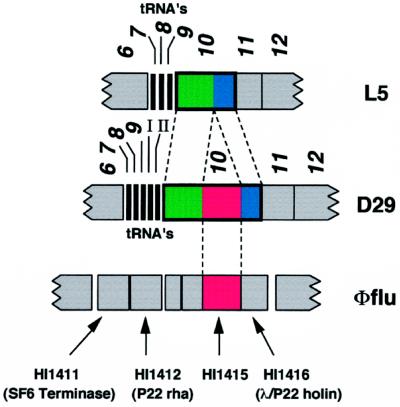
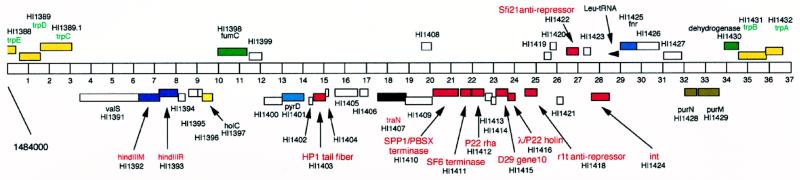
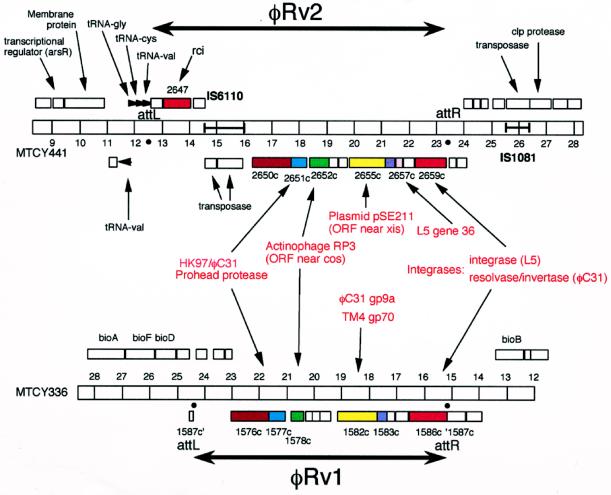
We report DNA and predicted protein sequence similarities, implying homology, among genes of double-stranded DNA (dsDNA) bacteriophages and prophages spanning a broad phylogenetic range of host bacteria. The sequence matches reported here establish genetic connections, not always direct, among the lambdoid phages of Escherichia coli, phage phiC31 of Streptomyces, phages of Mycobacterium, a previously unrecognized cryptic prophage, phiflu, in the Haemophilus influenzae genome, and two small prophage-like elements, phiRv1 and phiRv2, in the genome of Mycobacterium tuberculosis. The results imply that these phage genes, and very possibly all of the dsDNA tailed phages, share common ancestry. We propose a model for the genetic structure and dynamics of the global phage population in which all dsDNA phage genomes are mosaics with access, by horizontal exchange, to a large common genetic pool but in which access to the gene pool is not uniform for all phage.
Figures
References
-
- Coetzee J. In: Phage Ecology. Goval S M, Gerba C, Bitton G, editors. New York: Wiley; 1987. pp. 45–85.
-
- Ackermann H-W. Arch Virol. 1996;141:209–218. - PubMed
-
- Bergh Ø, Børsheim Y, Bratbak G, Heldal M. Nature (London) 1989;340:467–468. - PubMed
-
- Casjens S R, Hatfull G F, Hendrix R W. Semin Virol. 1992;3:383–397.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
