

Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency
- PMID: 10051646
- PMCID: PMC26788
- DOI: 10.1073/pnas.96.5.2356
Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency
Abstract
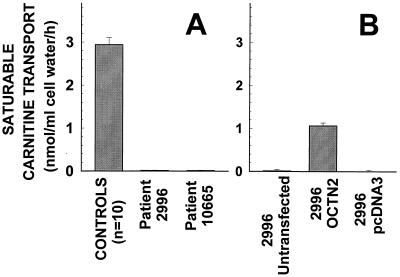
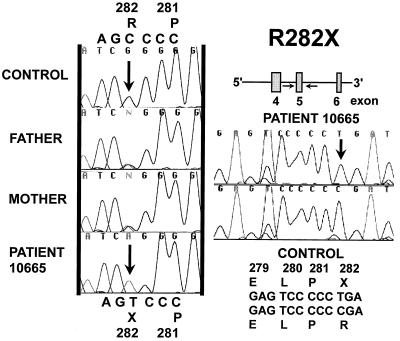
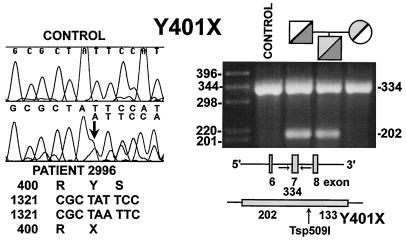
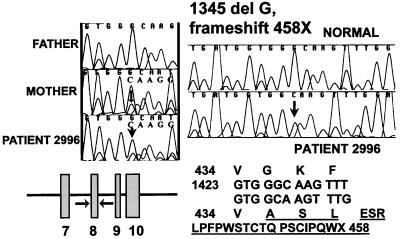
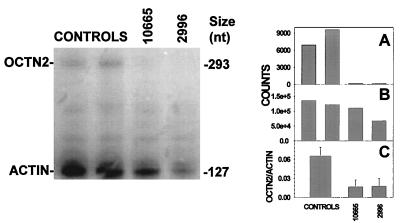
Primary carnitine deficiency is an autosomal recessive disorder of fatty acid oxidation caused by defective carnitine transport. This disease presents early in life with hypoketotic hypoglycemia or later in life with skeletal myopathy or cardiomyopathy. The gene for this condition maps to 5q31.2-32 and OCTN2, an organic cation/carnitine transporter, also maps to the same chromosomal region. Here we test the causative role of OCTN2 in primary carnitine deficiency by searching for mutations in this gene in affected patients. Fibroblasts from patients with primary carnitine deficiency lacked mediated carnitine transport. Transfection of patient's fibroblasts with the OCTN2 cDNA partially restored carnitine transport. Sequencing of the OCTN2 gene revealed different mutations in two unrelated patients. The first patient was homozygous (and both parents heterozygous) for a single base pair substitution converting the codon for Arg-282 to a STOP codon (R282X). The second patient was a compound heterozygote for a paternal 1-bp insertion producing a STOP codon (Y401X) and a maternal 1-bp deletion that produced a frameshift creating a subsequent STOP codon (458X). These mutations decreased the levels of mature OCTN2 mRNA and resulted in nonfunctional transporters, confirming that defects in the organic cation/carnitine transporter OCTN2 are responsible for primary carnitine deficiency.
Figures
References
-
- Roe C R, Coates P M. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 1501–1533.
-
- Pons, R. & De Vivo, D. C. (1995) J. Child Neurol. 10, Suppl. 2, 2S8–2S24. - PubMed
-
- Engel A G, Angelini C. Science. 1973;179:899–902. - PubMed
-
- Treem W R, Stanley C A, Finegold D N, Hale D E, Coates P M. N Engl J Med. 1988;319:1331–1336. - PubMed
-
- Eriksson B O, Gustafson B, Lindstedt S, Nordin I. J Inherited Metab Dis. 1989;12:108–111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
