

Coexistence of mitochondrial DNA and beta myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure
- PMID: 10065021
- PMCID: PMC1728869
- DOI: 10.1136/hrt.80.6.548
Coexistence of mitochondrial DNA and beta myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure
Erratum in
- Heart 1999 Mar;81(3):330
Abstract
Objective: To investigate the possible coexistence of mitochondrial DNA (mtDNA) mutations in patients with beta myosin heavy chain (beta MHC) linked hypertrophic cardiomyopathy (HCM) who develop congestive heart failure.
Design: Molecular analysis of beta MHC and mtDNA gene defects in patients with HCM.
Setting: Cardiovascular molecular diagnostic and heart transplantation reference centre in north Italy.
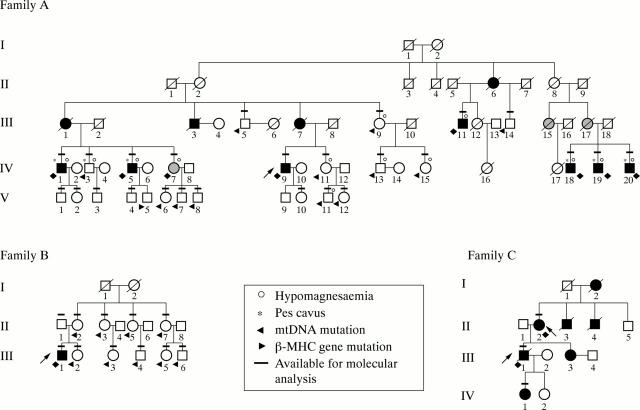
Patients: Four patients with HCM who underwent heart transplantation for end stage heart failure, and after pedigree analysis of 60 relatives, eight additional affected patients and 27 unaffected relatives. A total of 111 unrelated healthy adult volunteers served as controls. Disease controls included an additional 27 patients with HCM and 102 with dilated cardiomyopathy.
Intervention: Molecular analysis of DNA from myocardial and skeletal muscle tissue and from peripheral blood specimens.
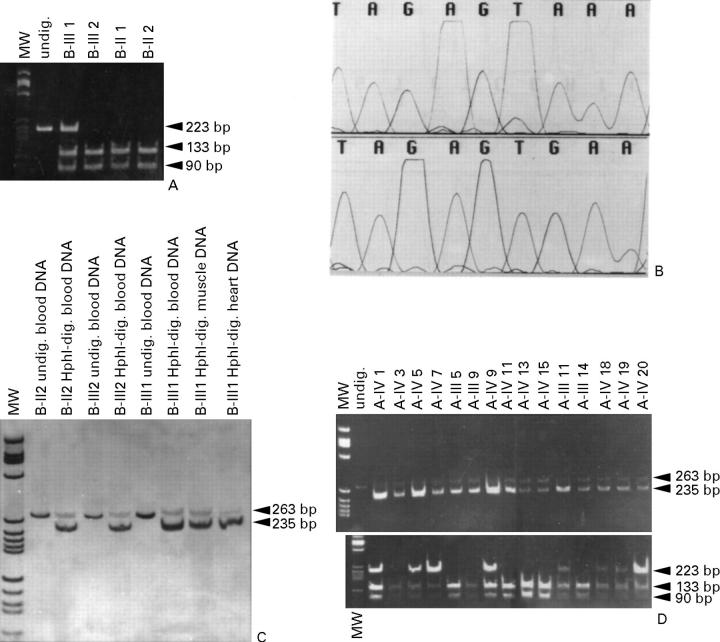
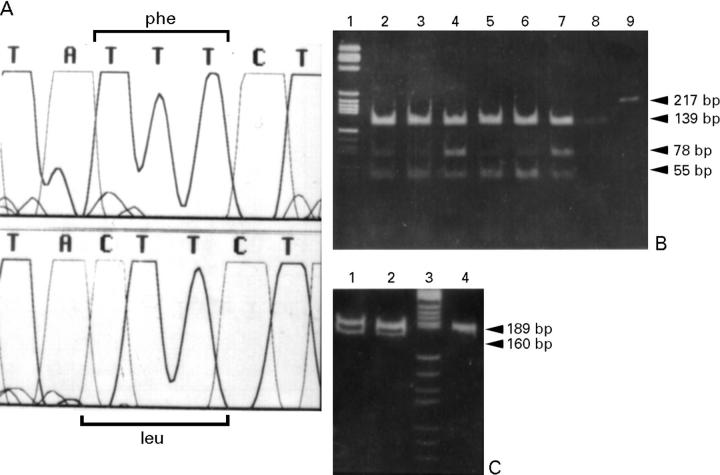
Main outcome measures: Screening for mutations in beta MHC (exons 3-23) and mtDNA tRNA (n = 22) genes with denaturing gradient gel electrophoresis or single strand conformational polymorphism followed by automated DNA sequencing.
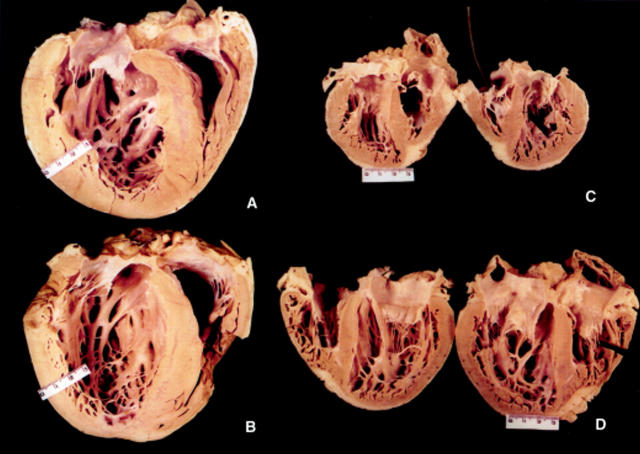
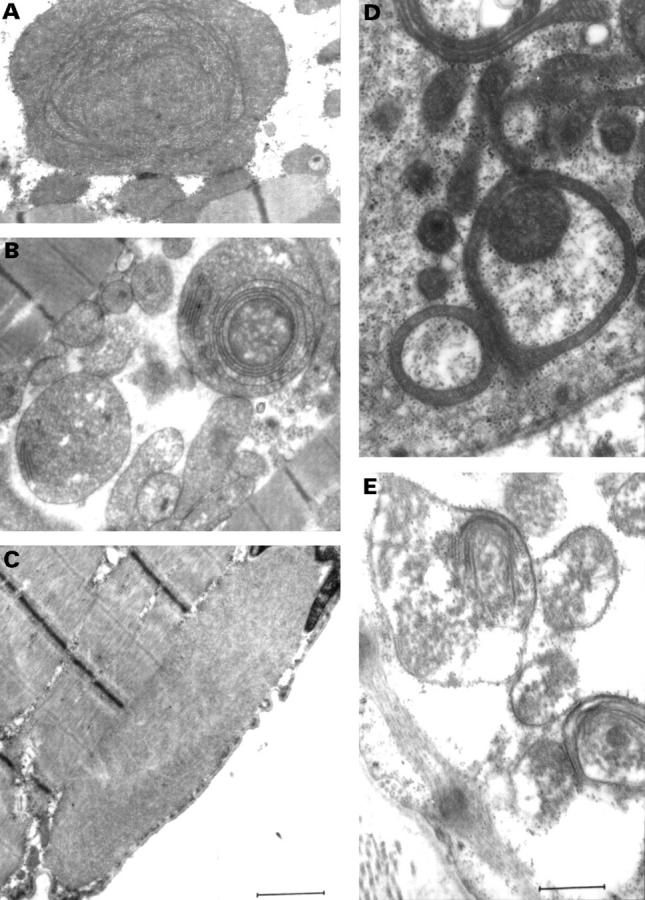
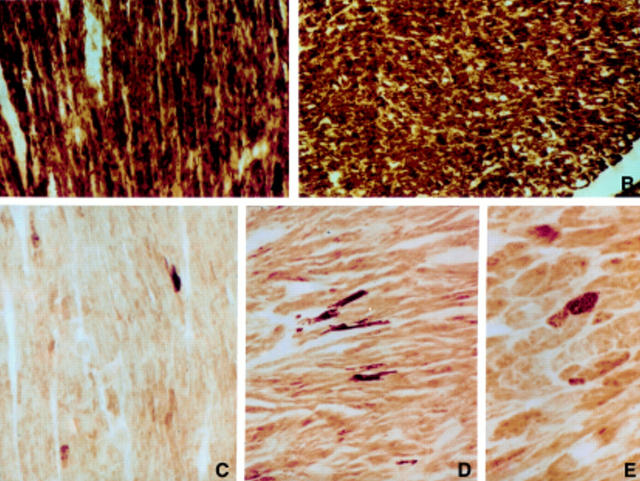
Results: One proband (kindred A) (plus seven affected relatives) had arginine 249 glutamine (Arg249Gln) beta MHC and heteroplasmic mtDNA tRNAIle A4300G mutations. Another unrelated patient (kindred B) with sporadic HCM had identical mutations. The remaining two patients (kindred C), a mother and son, had a novel beta MHC mutation (lysine 450 glutamic acid) (Lys450Glu) and a heteroplasmic missense (T9957C, phenylalanine (Phe)-->leucine (Leu)) mtDNA mutation in subunit III of the cytochrome C oxidase gene. The amount of mutant mtDNA was higher in the myocardium than in skeletal muscle or peripheral blood and in affected patients than in asymptomatic relatives. Mutations were absent in the controls. Pathological and biochemical characteristics of patients with mutations Arg249Gln plus A4300G (kindreds A and B) were identical, but different from those of the two patients with Lys450Glu plus T9957C(Phe-->Leu) mutations (kindred C). Cytochrome C oxidase activity and histoenzymatic staining were severely decreased in the two patients in kindreds A and B, but were unaffected in the two in kindred C.
Conclusions: beta MHC gene and mtDNA mutations may coexist in patients with HCM and end stage congestive heart failure. Although beta MHC gene mutations seem to be the true determinants of HCM, both mtDNA mutations in these patients have known prerequisites for pathogenicity. Coexistence of other genetic abnormalities in beta MHC linked HCM, such as mtDNA mutations, may contribute to variable phenotypic expression and explain the heterogeneous behaviour of HCM.
Figures
Similar articles
-
The cardiac beta-myosin heavy chain gene is not the predominant gene for hypertrophic cardiomyopathy in the Finnish population.J Am Coll Cardiol. 1998 Nov 15;32(6):1709-16. doi: 10.1016/s0735-1097(98)00448-3. J Am Coll Cardiol. 1998. PMID: 9822100
-
Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy.Proc Natl Acad Sci U S A. 1993 May 1;90(9):3993-7. doi: 10.1073/pnas.90.9.3993. Proc Natl Acad Sci U S A. 1993. PMID: 8483915 Free PMC article.
-
Clinical and prognostic evaluation of familial hypertrophic cardiomyopathy in two South African families with different cardiac beta myosin heavy chain gene mutations.Br Heart J. 1995 Jul;74(1):40-6. doi: 10.1136/hrt.74.1.40. Br Heart J. 1995. PMID: 7662452 Free PMC article.
-
Molecular basis of hypertrophic and dilated cardiomyopathy.Tex Heart Inst J. 1994;21(1):6-15. Tex Heart Inst J. 1994. PMID: 8180512 Free PMC article. Review.
-
Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives.J Am Coll Cardiol. 2012 Aug 21;60(8):705-15. doi: 10.1016/j.jacc.2012.02.068. Epub 2012 Jul 11. J Am Coll Cardiol. 2012. PMID: 22796258 Review.
Cited by
-
Whole mitochondrial genome analysis in Chinese patients with keratoconus.Mol Vis. 2021 May 8;27:270-282. eCollection 2021. Mol Vis. 2021. PMID: 34012229 Free PMC article.
-
Identification of a new missense mutation in the mtDNA of hereditary hypertrophic, but not dilated cardiomyopathic hamsters.Mol Cell Biochem. 2003 Oct;252(1-2):73-81. doi: 10.1023/a:1025542731335. Mol Cell Biochem. 2003. PMID: 14577578
-
Mavacamten inhibits myosin activity by stabilising the myosin interacting-heads motif and stalling motor force generation.bioRxiv [Preprint]. 2025 Feb 17:2025.02.12.637875. doi: 10.1101/2025.02.12.637875. bioRxiv. 2025. PMID: 39990378 Free PMC article. Preprint.
-
Biochemical and molecular basis for mitochondrial cardiomyopathy in neonates and children.J Inherit Metab Dis. 2000 Sep;23(6):625-33. doi: 10.1023/a:1005638231195. J Inherit Metab Dis. 2000. PMID: 11032337
-
Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene.J Med Genet. 2006 Oct;43(10):829-32. doi: 10.1136/jmg.2005.040329. Epub 2006 May 5. J Med Genet. 2006. PMID: 16679492 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials