

Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease
- PMID: 10068649
- PMCID: PMC4269326
Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease
Abstract
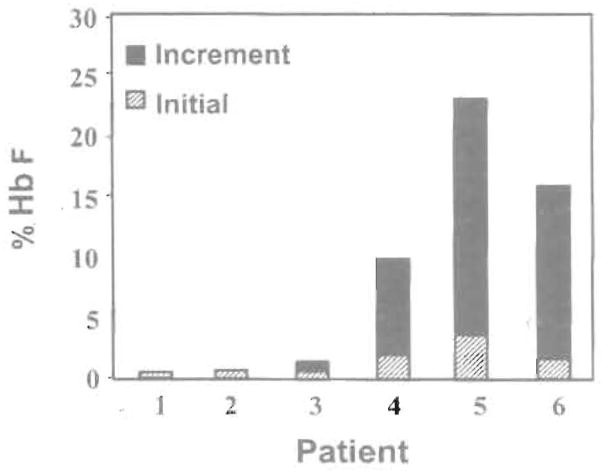
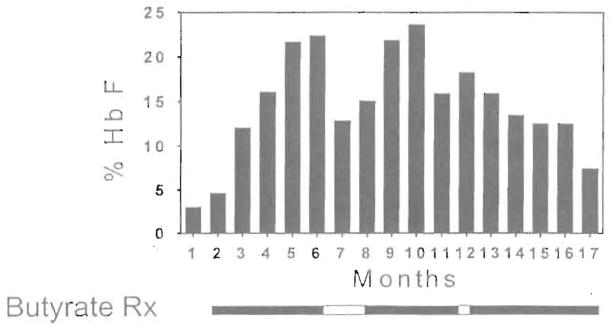
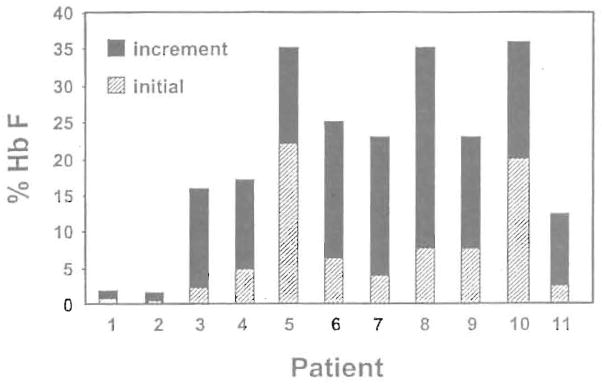
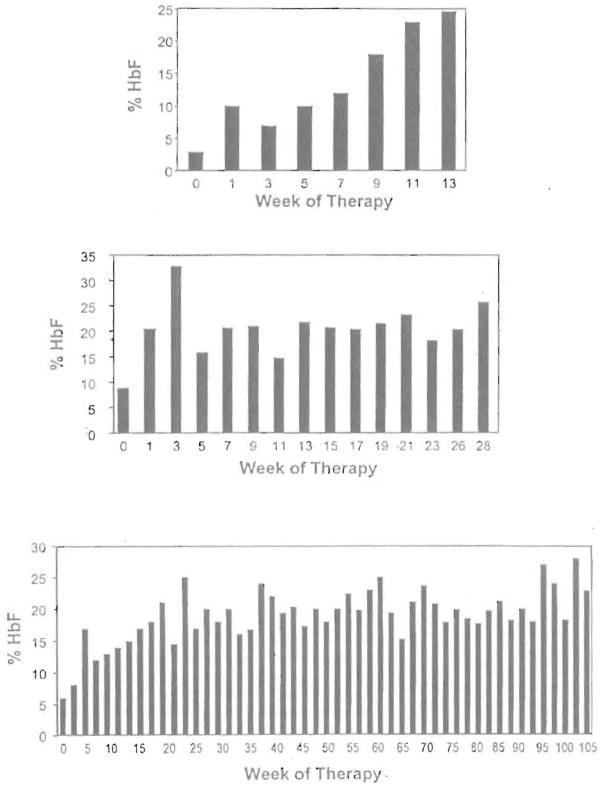
High levels of fetal hemoglobin (Hb F) protect from many of the complications of sickle cell disease and lead to improved survival. Butyrate and other short chain fatty acids were previously shown to increase Hb F production in erythroid cells in vitro and in animal models in vivo. However, butyrates are also known to inhibit the proliferation of many cell types, including erythroid cells. Experience with the use of butyrate in animal models and in early clinical trials demonstrated that the Hb F response may be lost after prolonged administration of high doses of butyrate. We hypothesized that this loss of response may be a result of the antiproliferative effects of butyrate. We designed a regimen consisting of intermittent or pulse therapy in which butyrate was administered for 4 days followed by 10 to 24 days with no drug exposure. This pulse regimen induced fetal globin gene expression in 9 of 11 patients. The mean Hb F in this group increased from 7.2% to 21.0% (P <.002) after intermittent butyrate therapy for a mean duration of 29.9 weeks. This was associated with a parallel increase in the number of F cells and F reticulocytes. The total hemoglobin levels also increased from a mean of 7.8 g/dL to a mean of 8.8 g/dL (P <.006). The increased levels of Hb F were sustained in all responders, including 1 patient who has been on pulse butyrate therapy for more than 28 months. This regimen, which resulted in a marked and sustained increase in Hb F levels in more than two thirds of the adult sickle cell patients enrolled in this study, was well tolerated without adverse side effects. These encouraging results require confirmation along with an appropriate evaluation of clinical outcomes in a larger number of patients with sickle cell disease.
Figures
Comment in
-
Induction of fetal hemoglobin in sickle cell disease.Blood. 1999 Mar 15;93(6):1787-9. Blood. 1999. PMID: 10068648 Review. No abstract available.
References
-
- Conley CL. Sickle cell anemia—The first molecular disease. In: Wintrobe MM, editor. Blood, Pure and Eloquent: A Story of Discovery, of People, and of Ideas. New York, NY: McGraw-Hill; 1980. p. 319.
-
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762. - PubMed
-
- Ingram VM. A specific chemical difference between the globins of normal human and sickle cell anemia haemoglobin. Nature. 1956;178:792. - PubMed
-
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization. Primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood. 1985;65:183. - PubMed
-
- Wood WG, Bunch C, Kelly S, Gunn Y, Breckon G. Control of haemoglobin switching by a developmental clock. Nature. 1985;313:320. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical