

Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations
- PMID: 10077612
- PMCID: PMC15870
- DOI: 10.1073/pnas.96.6.2919
Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations
Abstract
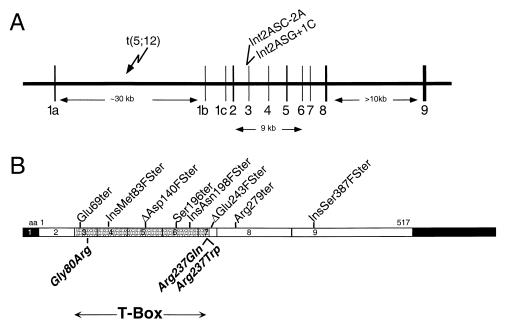
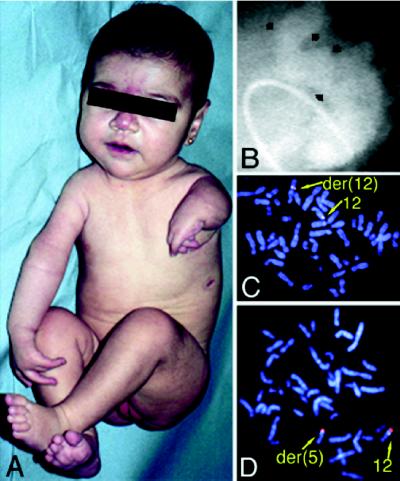
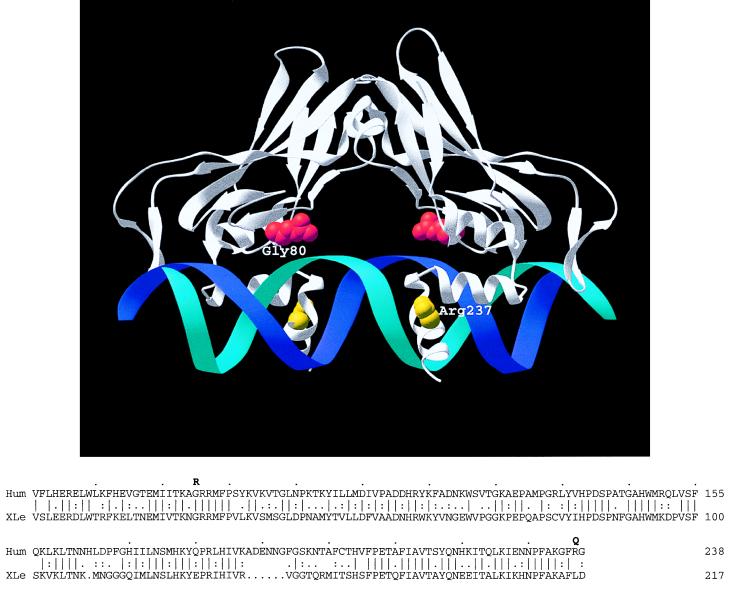
To better understand the role of TBX5, a T-box containing transcription factor in forelimb and heart development, we have studied the clinical features of Holt-Oram syndrome caused by 10 different TBX5 mutations. Defects predicted to create null alleles caused substantial abnormalities both in limb and heart. In contrast, missense mutations produced distinct phenotypes: Gly80Arg caused significant cardiac malformations but only minor skeletal abnormalities; and Arg237Gln and Arg237Trp caused extensive upper limb malformations but less significant cardiac abnormalities. Amino acids altered by missense mutations were located on the three-dimensional structure of a related T-box transcription factor, Xbra, bound to DNA. Residue 80 is highly conserved within T-box sequences that interact with the major groove of target DNA; residue 237 is located in the T-box domain that selectively binds to the minor groove of DNA. These structural data, taken together with the predominant cardiac or skeletal phenotype produced by each missense mutation, suggest that organ-specific gene activation by TBX5 is predicated on biophysical interactions with different target DNA sequences.
Figures
Comment in
-
Heart or hand? Unmasking the basis for specific Holt-Oram phenotypes.Proc Natl Acad Sci U S A. 1999 Mar 16;96(6):2577-8. doi: 10.1073/pnas.96.6.2577. Proc Natl Acad Sci U S A. 1999. PMID: 10077549 Free PMC article. No abstract available.
References
-
- Bollag R J, Siegfried Z, Cebra-Thomas J A, Garvey N, Davison E M, Silver L M. Nat Genet. 1994;7:383–389. - PubMed
-
- Holland P W, Koschorz B, Holland L Z, Herrmann B G. Development (Cambridge, UK) 1995;121:4283–4291. - PubMed
-
- Agulnik S I, Ruvinsky I, Silver L M. Genome. 1997;40:458–464. - PubMed
-
- Muller C W, Herrmann B G. Nature (London) 1997;389:884–888. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
