

Automated MAD and MIR structure solution
- PMID: 10089316
- PMCID: PMC2746121
- DOI: 10.1107/s0907444999000839
Automated MAD and MIR structure solution
Abstract
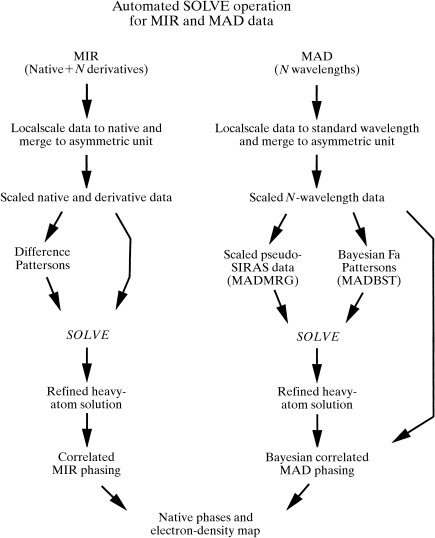
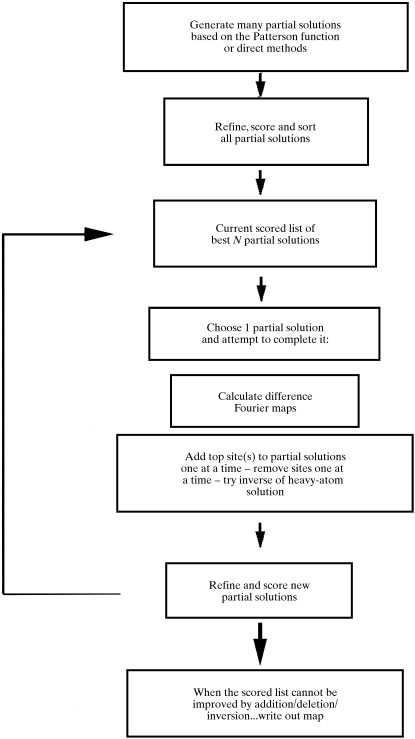
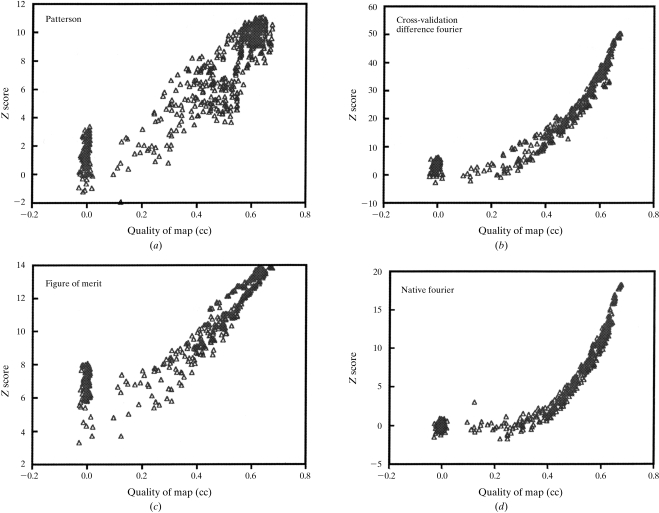
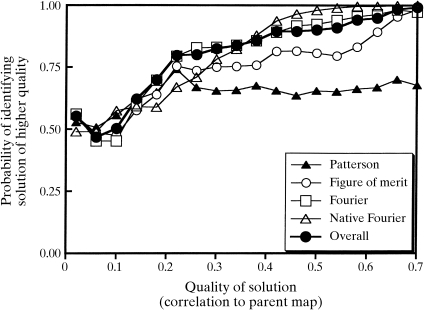
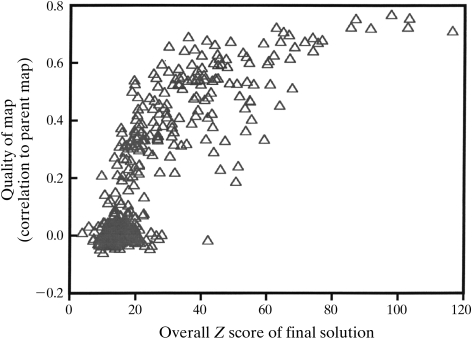
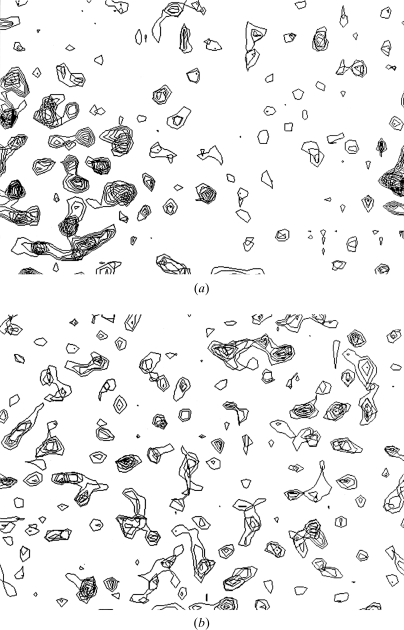
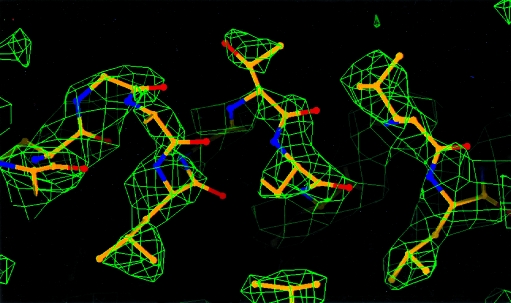
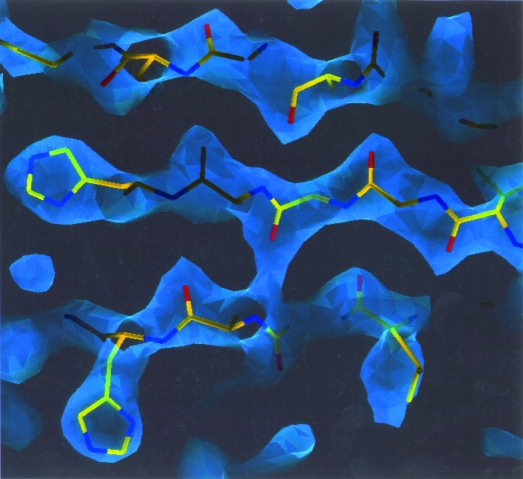
Obtaining an electron-density map from X-ray diffraction data can be difficult and time-consuming even after the data have been collected, largely because MIR and MAD structure determinations currently require many subjective evaluations of the qualities of trial heavy-atom partial structures before a correct heavy-atom solution is obtained. A set of criteria for evaluating the quality of heavy-atom partial solutions in macromolecular crystallography have been developed. These have allowed the conversion of the crystal structure-solution process into an optimization problem and have allowed its automation. The SOLVE software has been used to solve MAD data sets with as many as 52 selenium sites in the asymmetric unit. The automated structure-solution process developed is a major step towards the fully automated structure-determination, model-building and refinement procedure which is needed for genomic scale structure determinations.
Figures
References
-
- Abrahams, J. P., Leslie, A. G. W., Lutter, R. & Walker, J. E. (1994). Nature (London), 370, 621–628. - PubMed
-
- American Type Culture Collection (1992). Catalogue of Bacteria and Bacteriophages, 18th ed., pp. 271–272.
-
- Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer, E. F., Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T. & Tasumi, M. (1977). J. Mol. Biol.112, 535–542. - PubMed
-
- Blundell, T. L. & Johnson, L. N. (1976). Protein Crystallography, p. 368. New York: Academic Press.
-
- Buerger, M. J. (1970). Contemporary Crystallography. New York: McGraw-Hill.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
