

Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis
- PMID: 10097113
- PMCID: PMC22370
- DOI: 10.1073/pnas.96.7.3775
Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis
Abstract
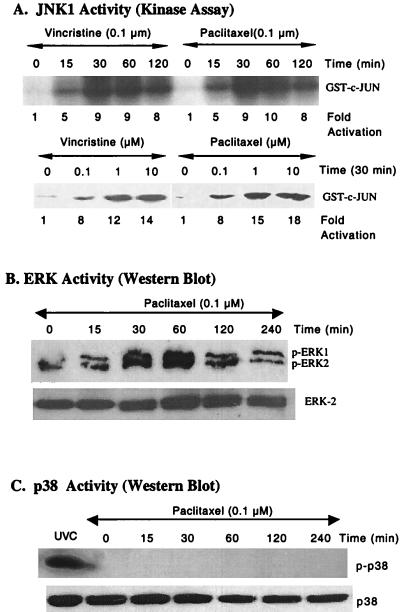
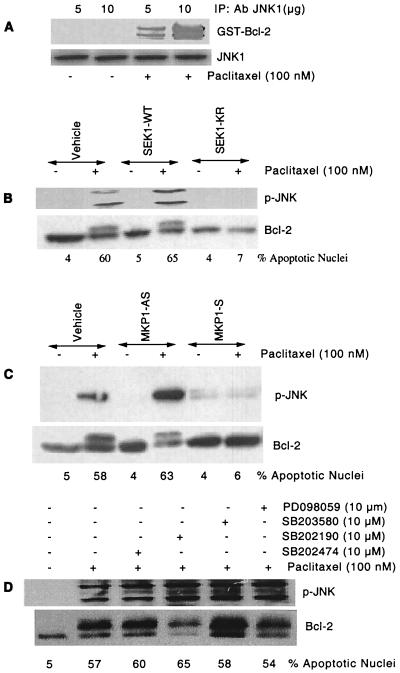
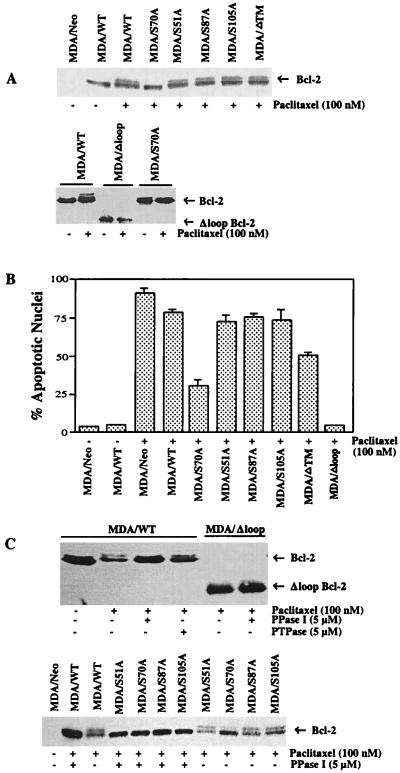
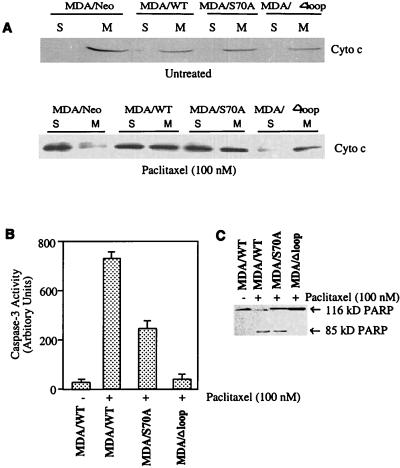
At high concentrations, the tubule poison paclitaxel is able to kill cancer cells that express Bcl-2; it inhibits the antiapoptotic activity of Bcl-2 by inducing its phosphorylation. To localize the site on Bcl-2 regulated by phosphorylation, mutant forms of Bcl-2 were constructed. Mutant forms of Bcl-2 with an alteration in serine at amino acid 70 (S70A) or with deletion of a 60-aa loop region between the alpha1 and alpha2 helices (Deltaloop Bcl-2, which also deletes amino acid 70) were unable to be phosphorylated by paclitaxel treatment of MDA-MB-231 cells into which the genes for the mutant proteins were transfected. The Deltaloop mutant completely inhibited paclitaxel-induced apoptosis. In cells expressing the S70A mutant, paclitaxel induced about one-third the level of apoptosis seen with wild-type Bcl-2. To evaluate the role of mitogen-activated protein kinases (MAPKs) in Bcl-2 phosphorylation, the activation of c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 was examined. Paclitaxel-induced apoptosis was associated with phosphorylation of Bcl-2 and activation of ERK and JNK MAPKs. If JNK activation was blocked by transfections with either a stress-activated protein kinase kinase dominant-negative (K-->R) gene (which prevents the activation of a kinase upstream of JNK) or MAPK phosphatase-1 gene (which dephosphorylates and inactivates JNK), Bcl-2 phosphorylation did not occur, and the cells were not killed by paclitaxel. By contrast, neither an ERK inhibitor (PD098059) nor p38 inhibitors (SB203580 and SB202190) had an effect on Bcl-2 phosphorylation. Thus, our data show that the antiapoptotic effects of Bcl-2 can be overcome by phosphorylation of Ser-70; forms of Bcl-2 lacking the loop region are much more effective at preventing apoptosis than wild-type Bcl-2 because they cannot be phosphorylated. JNK, but not ERK or p38 MAPK, appear to be involved in the phosphorylation of Bcl-2 induced by paclitaxel.
Figures
Similar articles
-
Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1.J Biol Chem. 1997 Oct 3;272(40):25238-42. doi: 10.1074/jbc.272.40.25238. J Biol Chem. 1997. PMID: 9312139
-
Involvement of Asp-Glu-Val-Asp-directed, caspase-mediated mitogen-activated protein kinase kinase 1 Cleavage, c-Jun N-terminal kinase activation, and subsequent Bcl-2 phosphorylation for paclitaxel-induced apoptosis in HL-60 cells.Mol Pharmacol. 2001 Feb;59(2):254-62. doi: 10.1124/mol.59.2.254. Mol Pharmacol. 2001. PMID: 11160861
-
A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation.Cancer Res. 2004 Mar 15;64(6):2134-42. doi: 10.1158/0008-5472.can-03-2296. Cancer Res. 2004. PMID: 15026354
-
Taxol induces apoptosis in cortical neurons by a mechanism independent of Bcl-2 phosphorylation.J Neurosci. 2001 Jul 1;21(13):4657-67. doi: 10.1523/JNEUROSCI.21-13-04657.2001. J Neurosci. 2001. PMID: 11425893 Free PMC article.
-
Serine protease inhibitor TPCK prevents Taxol-induced cell death and blocks c-Raf-1 and Bcl-2 phosphorylation in human breast carcinoma cells.Oncogene. 1999 Jun 10;18(23):3431-9. doi: 10.1038/sj.onc.1202685. Oncogene. 1999. PMID: 10376521
Cited by
-
Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription.J Exp Med. 1999 Jul 19;190(2):253-65. doi: 10.1084/jem.190.2.253. J Exp Med. 1999. PMID: 10432288 Free PMC article.
-
Bcl-xl does not have to bind Bax to protect T cells from death.J Exp Med. 2006 Dec 25;203(13):2953-61. doi: 10.1084/jem.20061151. Epub 2006 Dec 11. J Exp Med. 2006. PMID: 17158961 Free PMC article.
-
Microtubule-targeting drugs induce Bcl-2 phosphorylation and association with Pin1.Neoplasia. 2001 Jan-Feb;3(1):70-9. doi: 10.1038/sj.neo.7900131. Neoplasia. 2001. PMID: 11326318 Free PMC article.
-
Pin1 in neuronal apoptosis.Cell Cycle. 2007 Jun 1;6(11):1332-5. doi: 10.4161/cc.6.11.4316. Epub 2007 Jun 20. Cell Cycle. 2007. PMID: 17568190 Free PMC article.
-
Geridonin and paclitaxel act synergistically to inhibit the proliferation of gastric cancer cells through ROS-mediated regulation of the PTEN/PI3K/Akt pathway.Oncotarget. 2016 Nov 8;7(45):72990-73002. doi: 10.18632/oncotarget.12166. Oncotarget. 2016. PMID: 27659528 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
