

In autoimmune diabetes the transition from benign to pernicious insulitis requires an islet cell response to tumor necrosis factor alpha
- PMID: 10190896
- PMCID: PMC2193009
- DOI: 10.1084/jem.189.7.1053
In autoimmune diabetes the transition from benign to pernicious insulitis requires an islet cell response to tumor necrosis factor alpha
Abstract
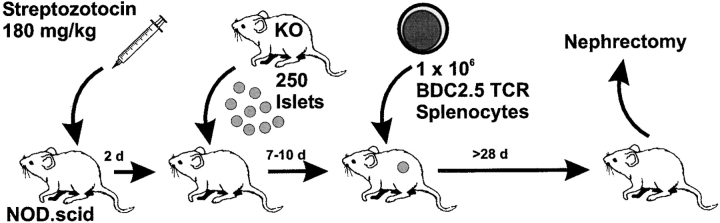
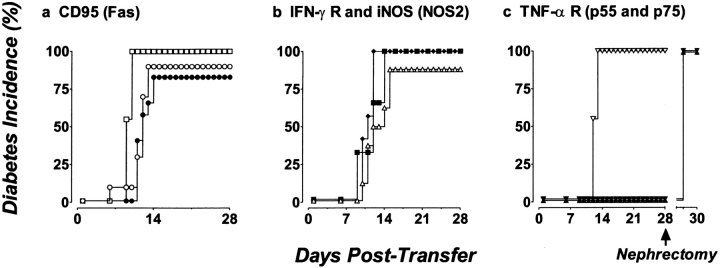
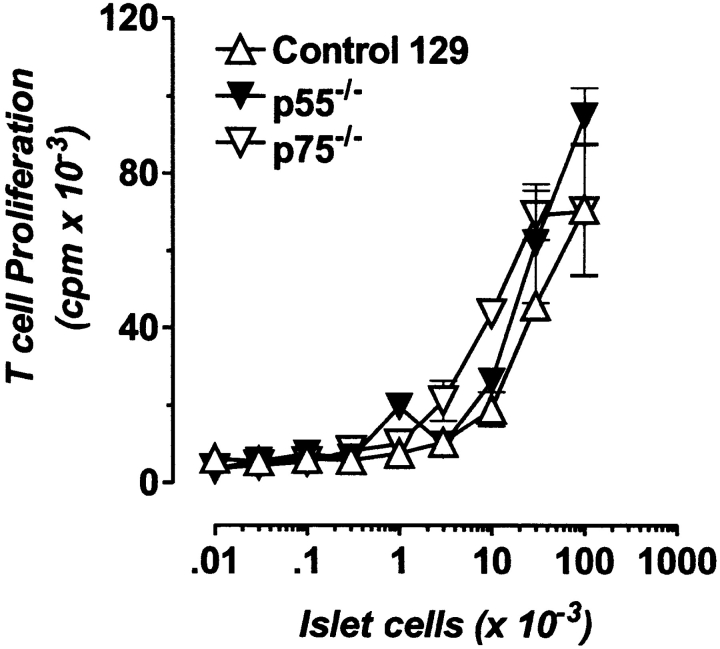
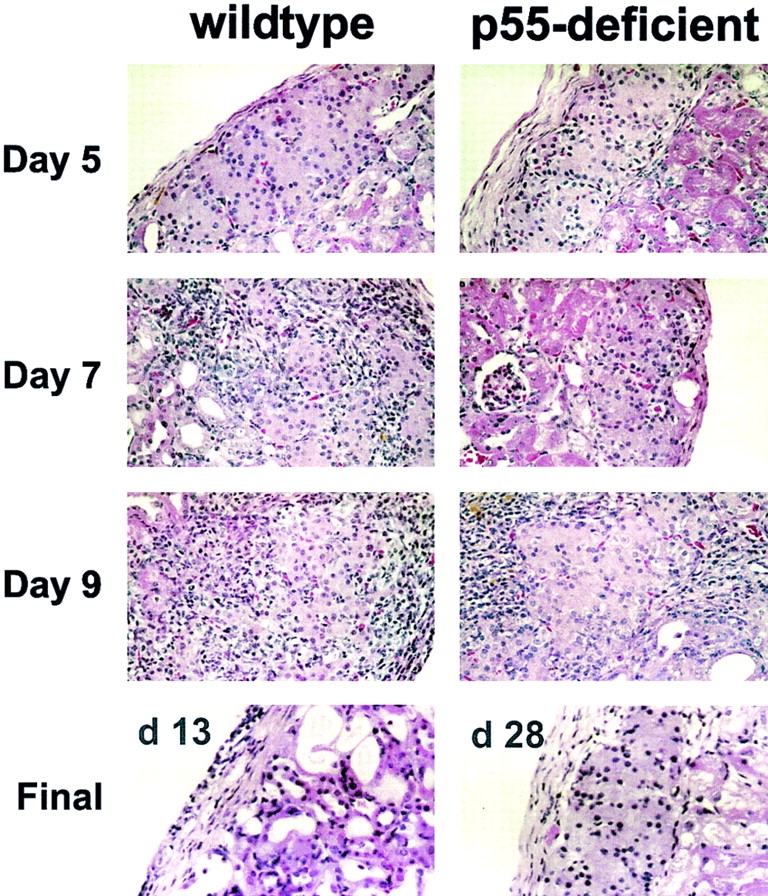
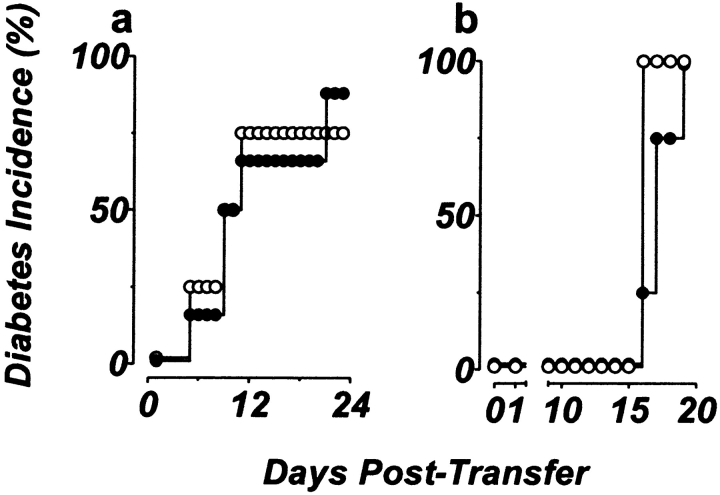
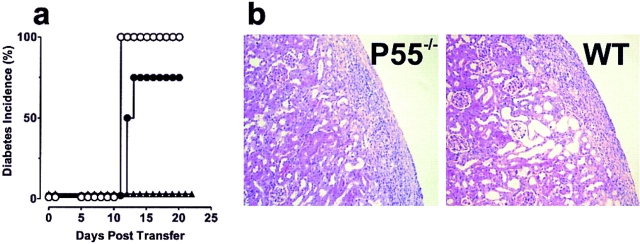
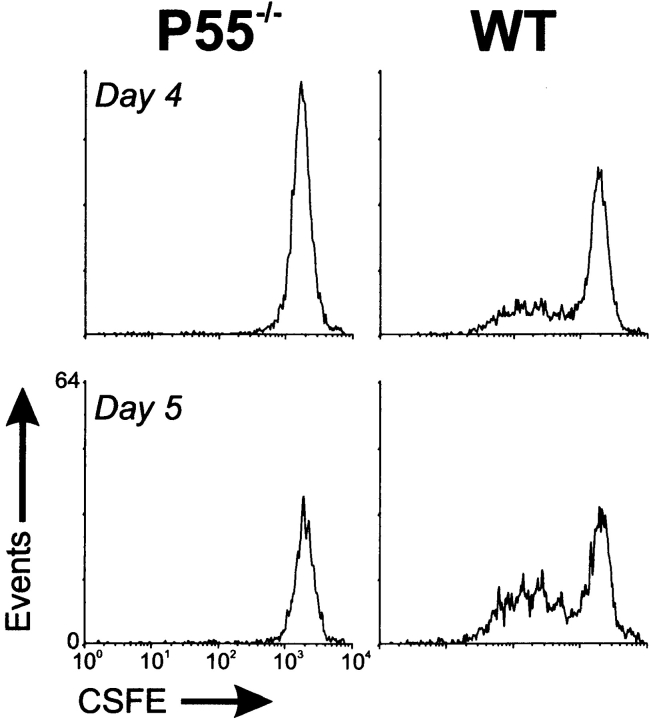
The islet-infiltrating and disease-causing leukocytes that are a hallmark of insulin-dependent diabetes mellitus produce and respond to a set of cytokine molecules. Of these, interleukin 1beta, tumor necrosis factor (TNF)-alpha, and interferon (IFN)-gamma are perhaps the most important. However, as pleiotropic molecules, they can impact the path leading to beta cell apoptosis and diabetes at multiple points. To understand how these cytokines influence both the formative and effector phases of insulitis, it is critical to determine their effects on the assorted cell types comprising the lesion: the effector T cells, antigen-presenting cells, vascular endothelium, and target islet tissue. Here, we report using nonobese diabetic chimeric mice harboring islets deficient in specific cytokine receptors or cytokine-induced effector molecules to assess how these compartmentalized loss-of-function mutations alter the events leading to diabetes. We found that islets deficient in Fas, IFN-gamma receptor, or inducible nitric oxide synthase had normal diabetes development; however, the specific lack of TNF- alpha receptor 1 (p55) afforded islets a profound protection from disease by altering the ability of islet-reactive, CD4(+) T cells to establish insulitis and subsequently destroy islet beta cells. These results argue that islet cells play a TNF-alpha-dependent role in their own demise.
Figures
References
-
- Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev. 1994;15:516–542. - PubMed
-
- Atkinson MA, MacLaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;331:1428–1436. - PubMed
-
- Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. - PubMed
-
- Charlton B, Bacelj A, Mandel TE. Administration of silica particles or anti-Lyt2 antibody prevents β-cell destruction in NOD mice given cyclophosphamide. Diabetes. 1988;37:930–935. - PubMed
-
- Miller BJ, Appel MC, O'Neil J, Wicker LS. Both the LYT-2+ and L3T4+T cell subsets are required for transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
