

Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation
- PMID: 10191337
- PMCID: PMC6782264
- DOI: 10.1523/JNEUROSCI.19-08-03248.1999
Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation
Abstract
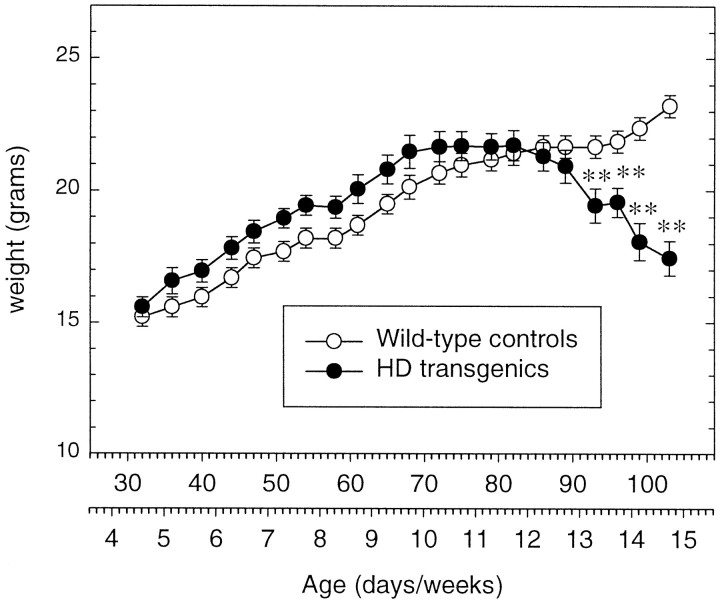
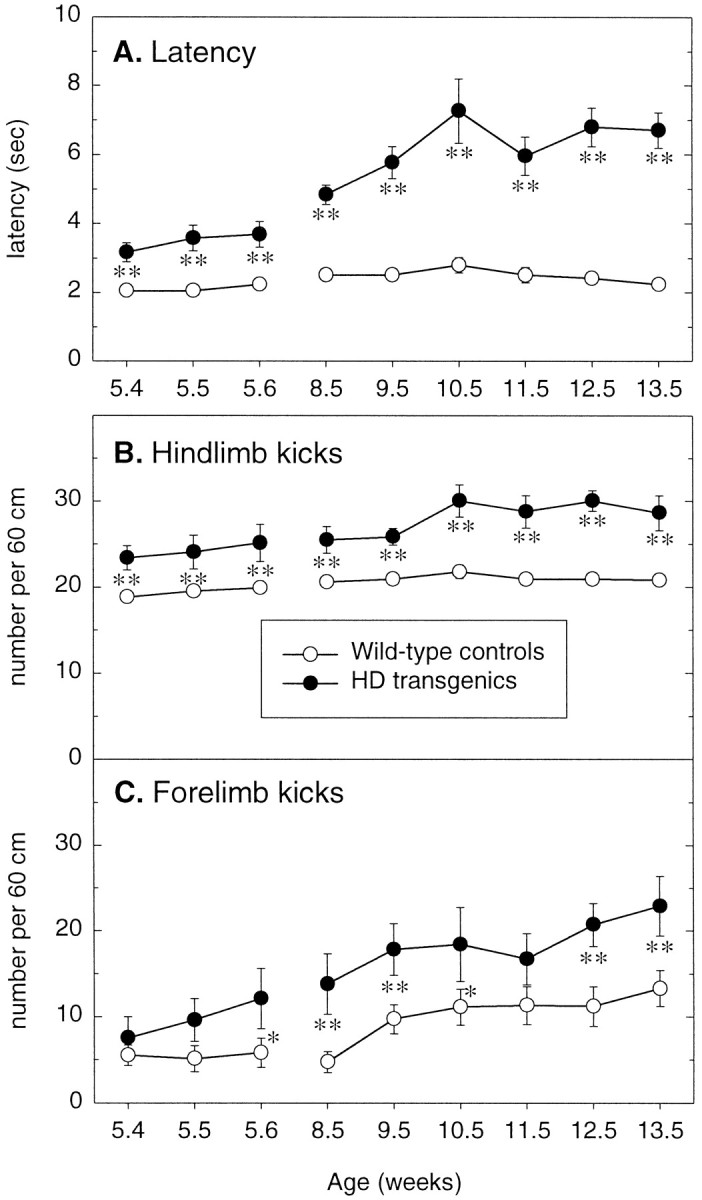
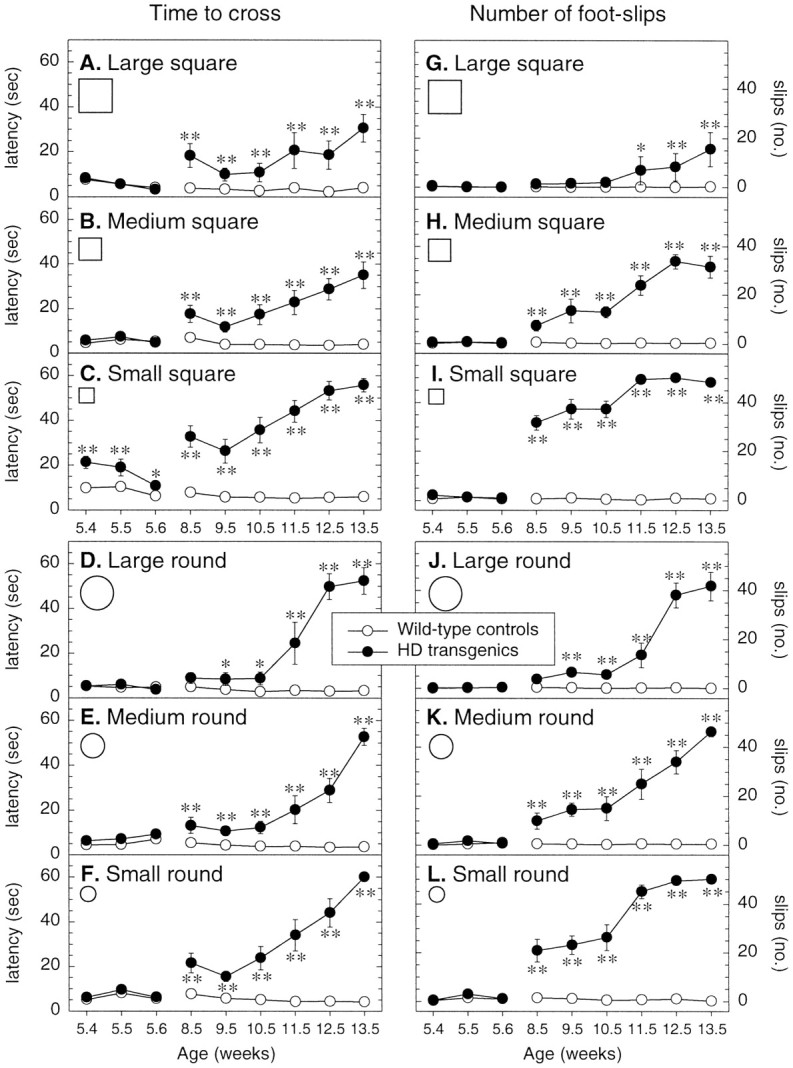
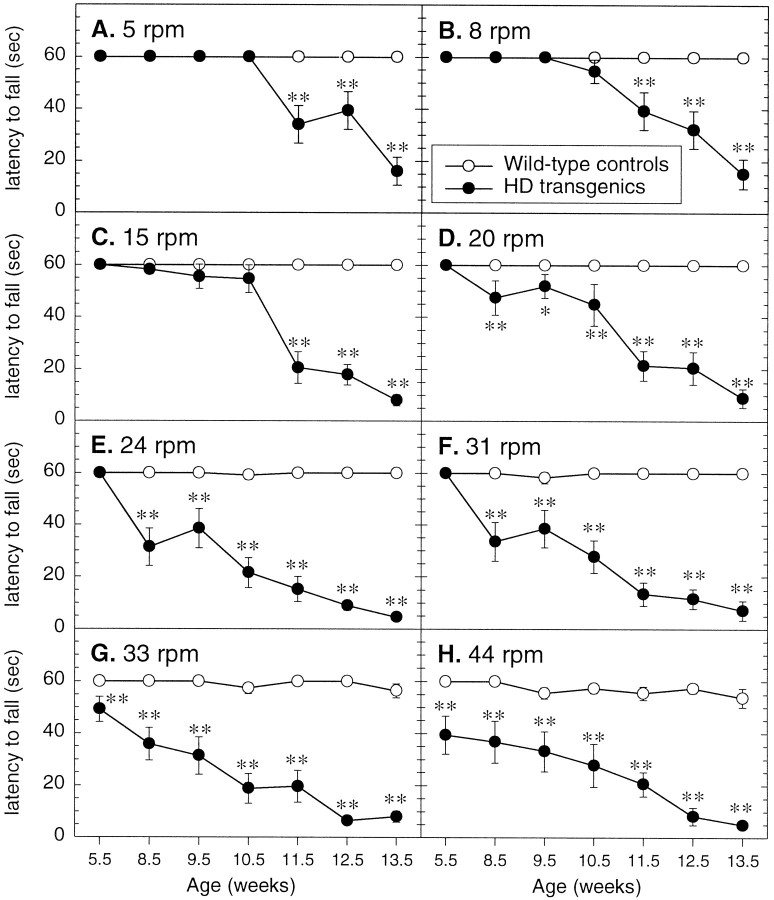
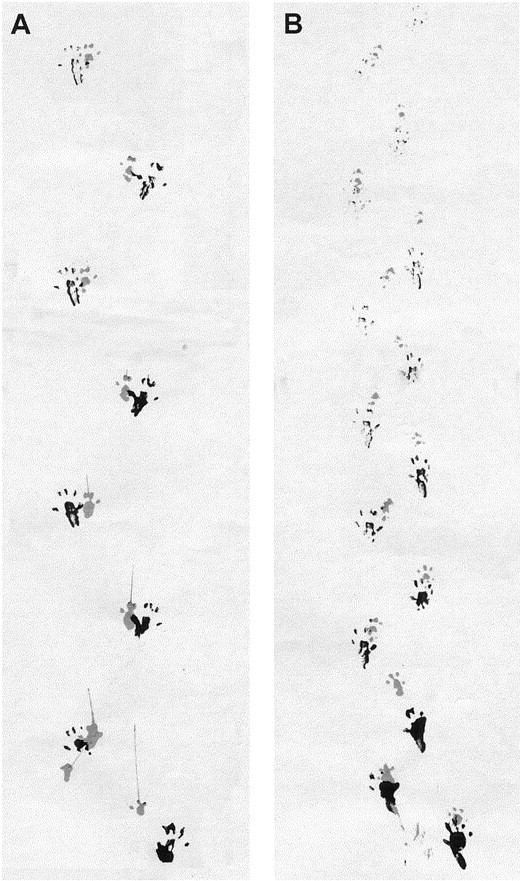
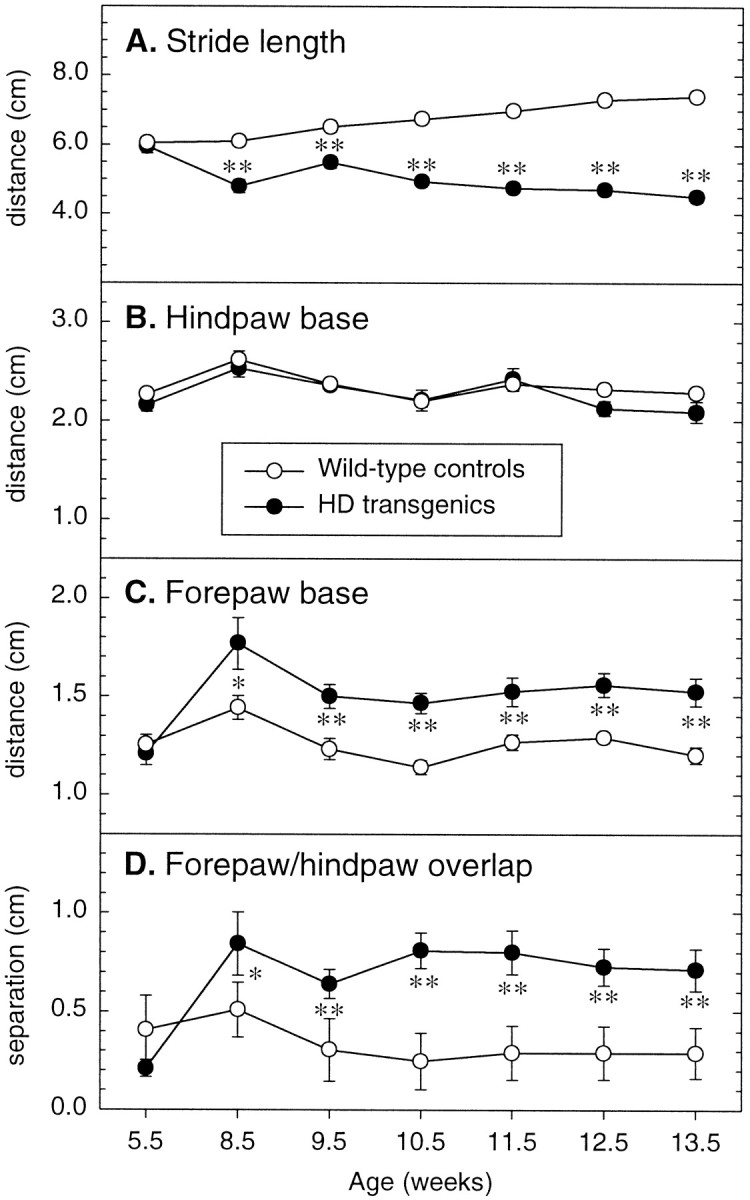
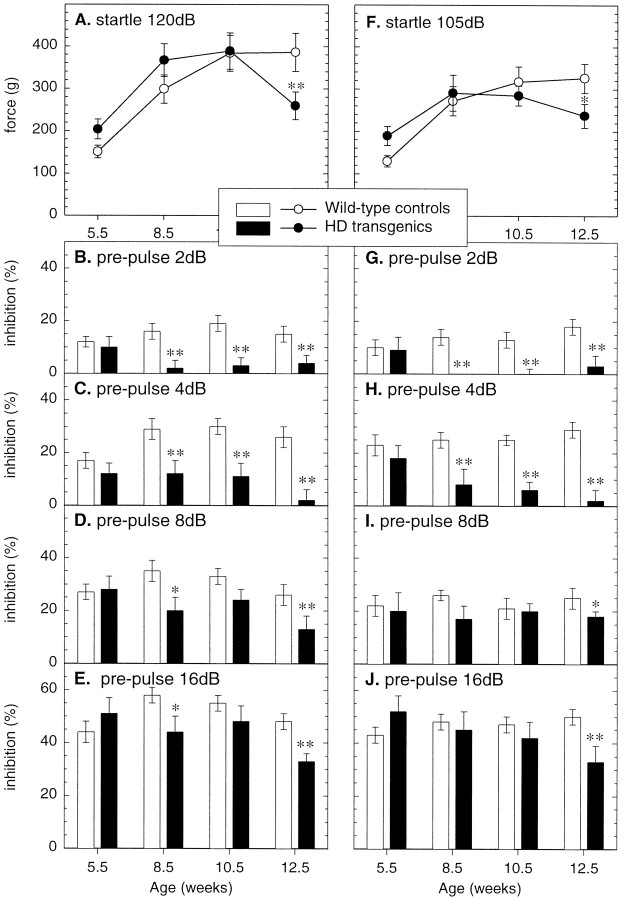
Transgenic mice expressing exon 1 of the human Huntington's disease (HD) gene carrying a 141-157 CAG repeat (line R6/2) develop a progressive neurological phenotype with motor symptoms resembling those seen in HD. We have characterized the motor deficits in R6/2 mice using a battery of behavioral tests selected to measure motor aspects of swimming, fore- and hindlimb coordination, balance, and sensorimotor gating [swimming tank, rotarod, raised beam, fore- and hindpaw footprinting, and acoustic startle/prepulse inhibition (PPI)]. Behavioral testing was performed on female hemizygotic R6/2 transgenic mice (n = 9) and female wild-type littermates (n = 22) between 5 and 14 weeks of age. Transgenic mice did not show an overt behavioral phenotype until around 8 weeks of age. However, as early as 5-6 weeks of age they had significant difficulty swimming, traversing the narrowest square (5 mm) raised beam, and maintaining balance on the rotarod at rotation speeds of 33-44 rpm. Furthermore, they showed significant impairment in prepulse inhibition (an impairment also seen in patients with HD). Between 8 and 15 weeks, R6/2 transgenic mice showed a progressive deterioration in performance on all of the motor tests. Thus R6/2 mice show measurable deficits in motor behavior that begin subtly and increase progressively until death. Our data support the use of R6/2 mice as a model of HD and indicate that they may be useful for evaluating therapeutic strategies for HD, particularly those aimed at reducing the severity of motor symptoms or slowing the course of the disease.
Figures
References
-
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–171. - PubMed
-
- Borlongan CV, Koutouzis TK, Freeman TB, Cahill DW, Sanberg PR. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington’s disease. Brain Res. 1995;697:254–257. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical