

Host-specific modulation of the selective constraints driving human immunodeficiency virus type 1 env gene evolution
- PMID: 10196271
- PMCID: PMC104154
- DOI: 10.1128/JVI.73.5.3764-3777.1999
Host-specific modulation of the selective constraints driving human immunodeficiency virus type 1 env gene evolution
Abstract
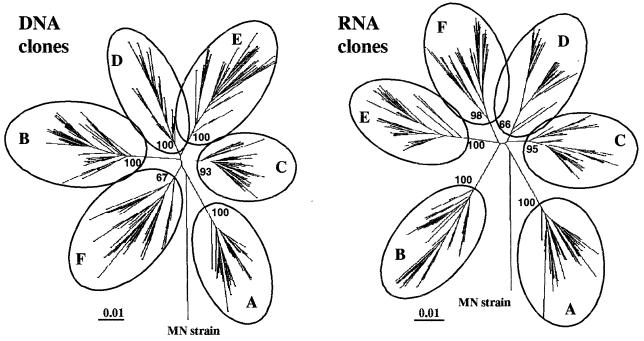
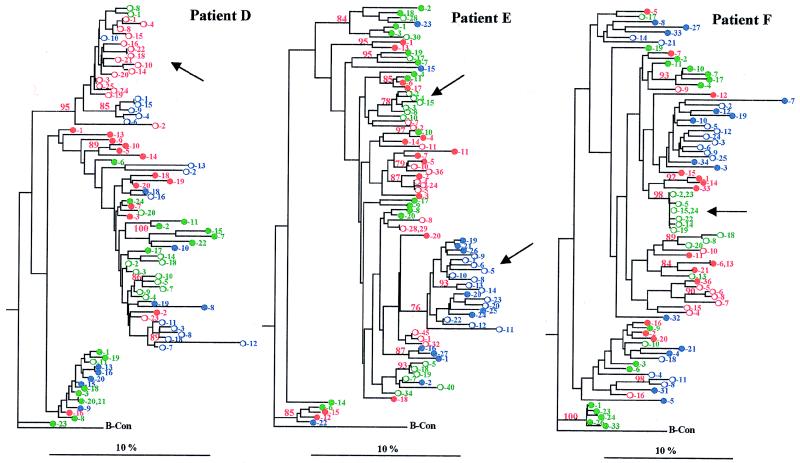
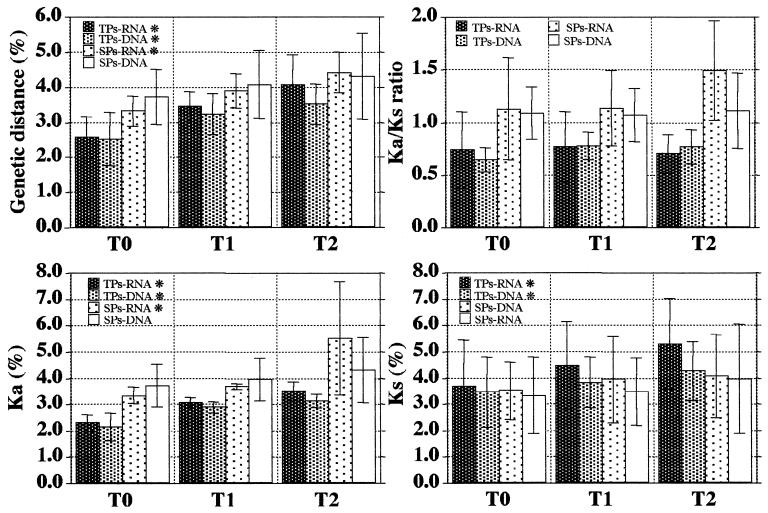
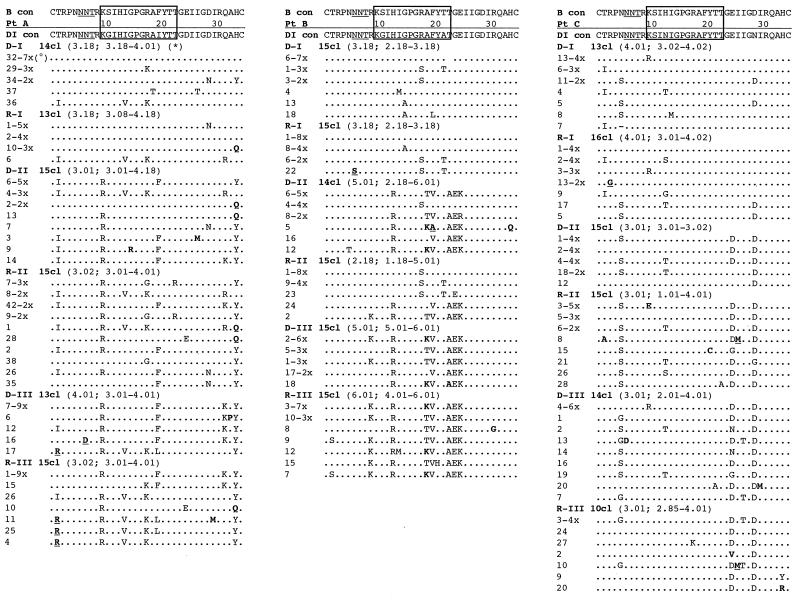
To address the evolution of human immunodeficiency virus type 1 (HIV-1) within a single host, we analyzed the HIV-1 C2-V5 env regions of both cell-free genomic-RNA- and proviral-DNA-derived clones. Sequential samples were collected over a period of 3 years from six untreated subjects (three typical progressors [TPs] and three slow progressors [SPs], all with a comparable length of infection except one. The evolutionary analysis of the C2-V5 env sequences performed on 506 molecular clones (253 RNA- and 253 DNA-derived sequences) highlighted a series of differences between TPs and SPs. In particular, (i) clonal sequences from SPs (DNA and RNA) showed lower nucleotide similarity than those from TPs (P = 0. 0001), (ii) DNA clones from SPs showed higher intra- and intersample nucleotide divergence than those from TPs (P < 0.05), (iii) higher host-selective pressure was generally detectable in SPs (DNA and RNA sequences), and (iv) the increase in the genetic distance of DNA and RNA sequences over time was paralleled by an increase in both synonymous (Ks) and nonsynonymous (Ka) substitutions in TPs but only in nonsynonymous substitutions in SPs. Several individual peculiarities of the HIV-1 evolutionary dynamics emerged when the V3, V4, and V5 env regions of both TPs and SPs were evaluated separately. These peculiarities, probably reflecting host-specific features of selective constraints and their continuous modulation, are documented by the dynamics of Ka/Ks ratios of hypervariable env domains.
Figures



References
-
- Bakhanashvili M, Hizi A. Fidelity of the reverse transcriptase of human immunodeficiency type 2. FEBS Lett. 1992;306:151–156. - PubMed
-
- Bakhanashvili M, Hizi A. The fidelity of the reverse transcriptases of human immunodeficiency viruses and murine leukemia virus exhibited by the mispair extension frequencies is sequence dependent and enzyme related. FEBS Lett. 1993;319:201–205. - PubMed
-
- Bebenek K, Abbotts J, Wilson S H, Kunkel T A. Error-prone polymerization by HIV-1 reverse transcriptase. Contribution of template-primer misalignment miscoding and termination probability to mutational hot spots. J Biol Chem. 1993;268:10324–10334. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
