

Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1
- PMID: 10198041
- PMCID: PMC25205
- DOI: 10.1091/mbc.10.4.833
Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1
Abstract
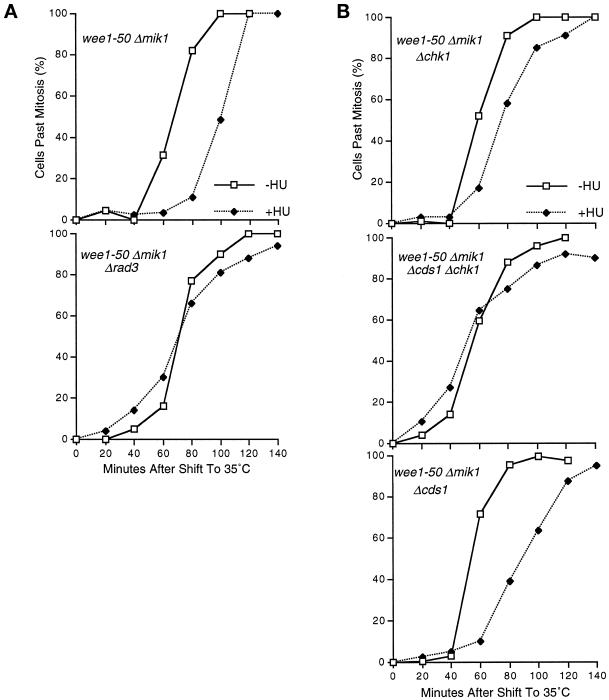
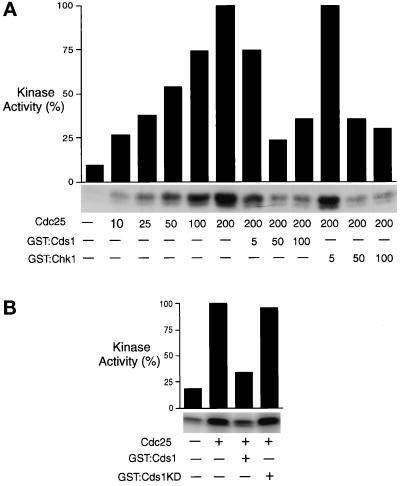
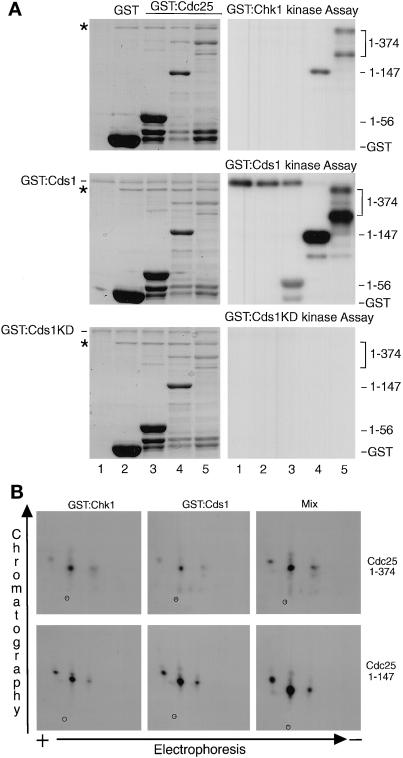
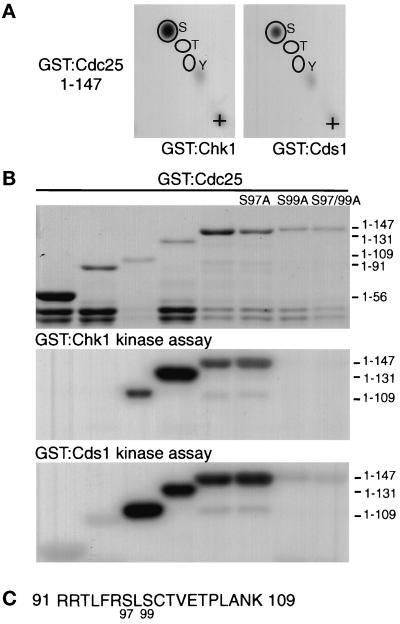
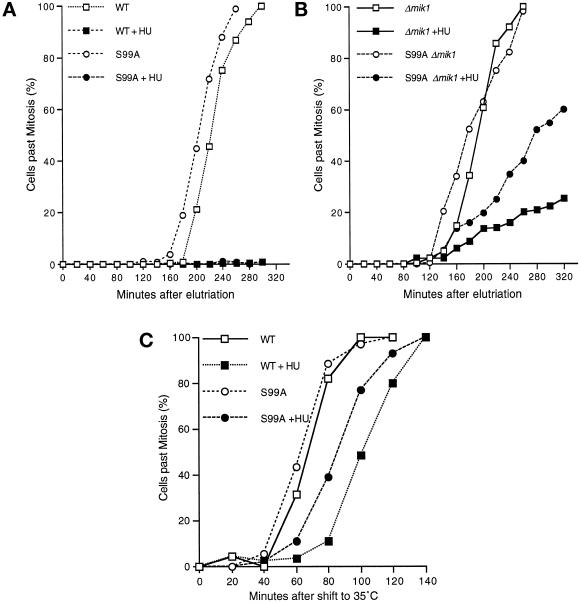
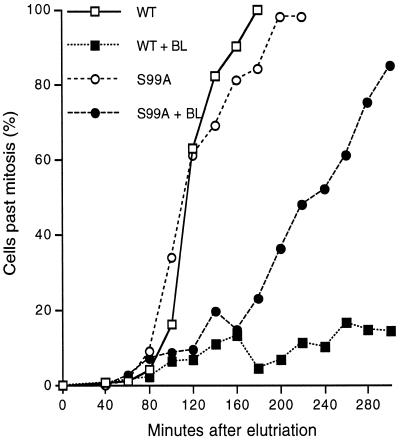
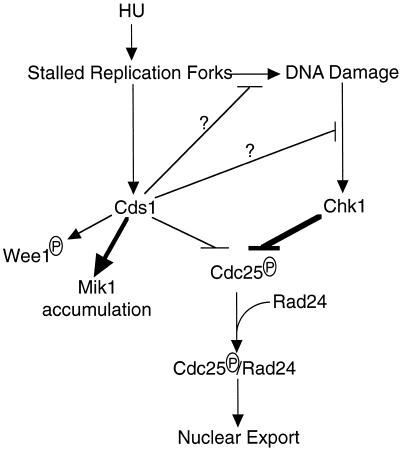
In the fission yeast Schizosaccharomyces pombe, the protein kinase Cds1 is activated by the S-M replication checkpoint that prevents mitosis when DNA is incompletely replicated. Cds1 is proposed to regulate Wee1 and Mik1, two tyrosine kinases that inhibit the mitotic kinase Cdc2. Here, we present evidence from in vivo and in vitro studies, which indicates that Cds1 also inhibits Cdc25, the phosphatase that activates Cdc2. In an in vivo assay that measures the rate at which Cdc25 catalyzes mitosis, Cds1 contributed to a mitotic delay imposed by the S-M replication checkpoint. Cds1 also inhibited Cdc25-dependent activation of Cdc2 in vitro. Chk1, a protein kinase that is required for the G2-M damage checkpoint that prevents mitosis while DNA is being repaired, also inhibited Cdc25 in the in vitro assay. In vitro, Cds1 and Chk1 phosphorylated Cdc25 predominantly on serine-99. The Cdc25 alanine-99 mutation partially impaired the S-M replication and G2-M damage checkpoints in vivo. Thus, Cds1 and Chk1 seem to act in different checkpoint responses to regulate Cdc25 by similar mechanisms.
Figures
References
-
- Blasina A, Van de Weyer I, Laus MC, Luyten WHML, Parker AE, McGowan CH. A human homolog of the checkpoint kinase Cds1 directly inhibits Cdc25. Curr Biol. 1999;9:1–10. - PubMed
-
- Boddy MN, Furnari B, Mondesert O, Russell P. Replication checkpoint enforced by kinases Cds1 and Chk1. Science. 1998;280:909–912. - PubMed
-
- Booher RN, Alfa CE, Hyams JS, Beach DH. The fission yeast cdc2/cdc13/suc1 protein kinase: regulation of catalytic activity and nuclear localization. Cell. 1989;58:485–497. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
