

Cellular activation triggered by the autosomal dominant polycystic kidney disease gene product PKD2
- PMID: 10207066
- PMCID: PMC84135
- DOI: 10.1128/MCB.19.5.3423
Cellular activation triggered by the autosomal dominant polycystic kidney disease gene product PKD2
Abstract
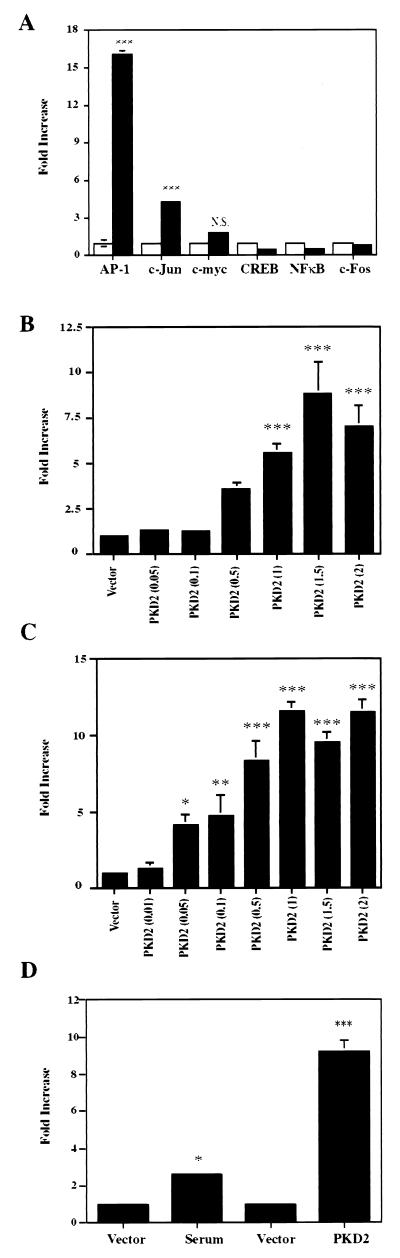
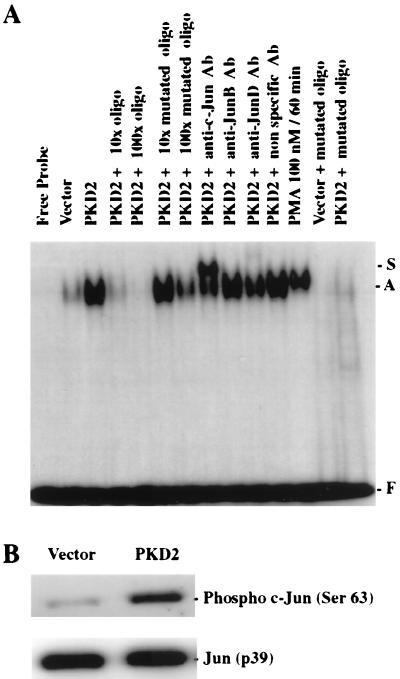
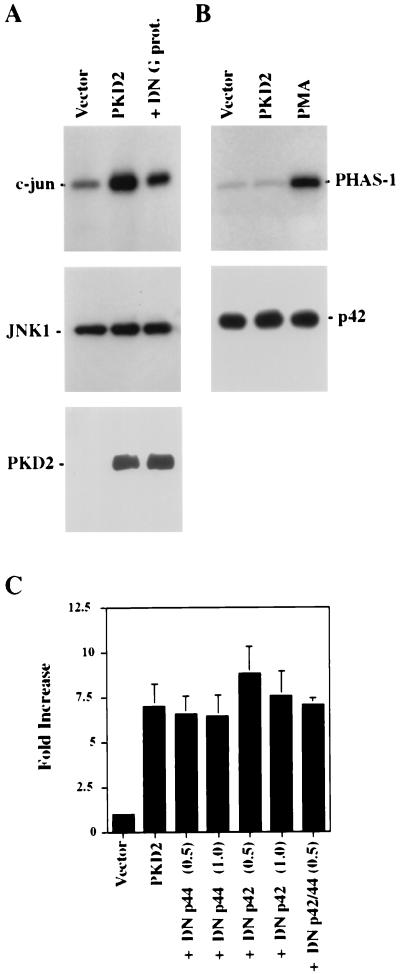
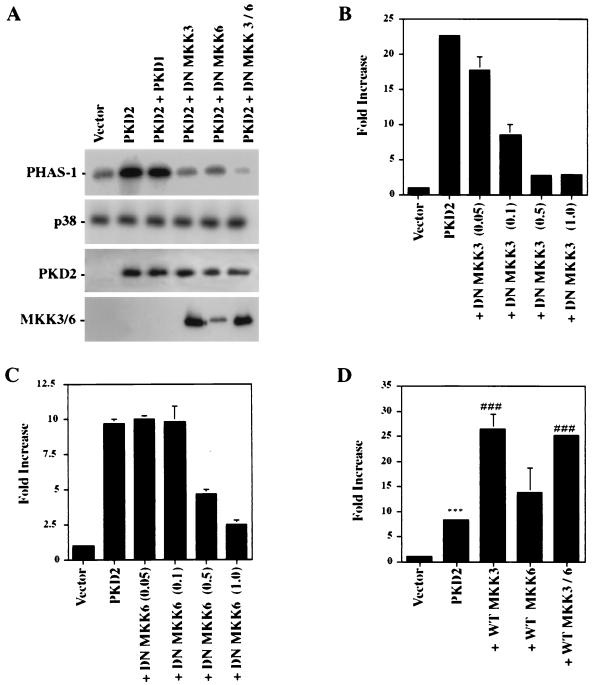
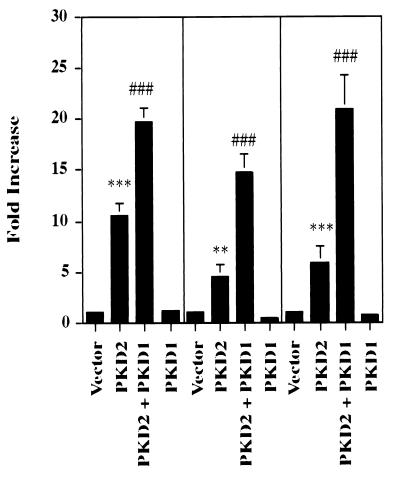
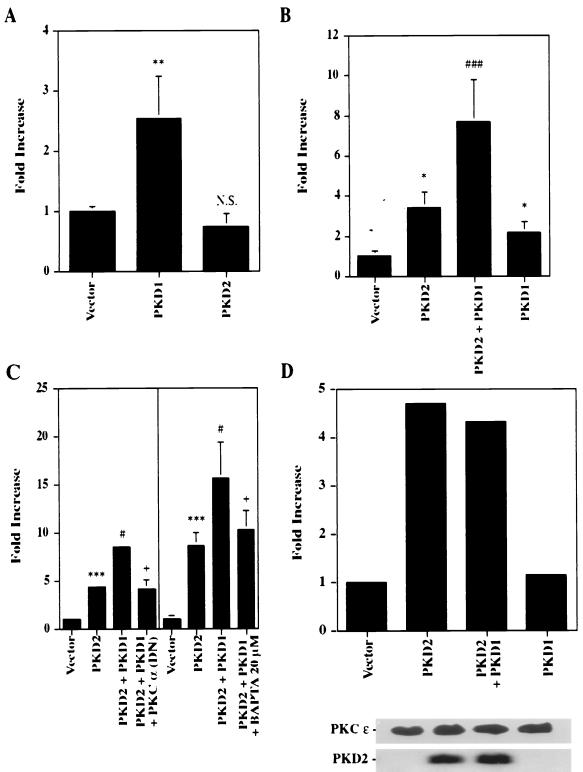
Autosomal dominant polycystic kidney disease (ADPKD) is caused by germ line mutations in at least three ADPKD genes. Two recently isolated ADPKD genes, PKD1 and PKD2, encode integral membrane proteins of unknown function. We found that PKD2 upregulated AP-1-dependent transcription in human embryonic kidney 293T cells. The PKD2-mediated AP-1 activity was dependent upon activation of the mitogen-activated protein kinases p38 and JNK1 and protein kinase C (PKC) epsilon, a calcium-independent PKC isozyme. Staurosporine, but not the calcium chelator BAPTA [1,2-bis(o-aminophenoxy)ethane-N,N,N', N'-tetraacetate], inhibited PKD2-mediated signaling, consistent with the involvement of a calcium-independent PKC isozyme. Coexpression of PKD2 with the interacting C terminus of PKD1 dramatically augmented PKD2-mediated AP-1 activation. The synergistic signaling between PKD1 and PKD2 involved the activation of two distinct PKC isozymes, PKC alpha and PKC epsilon, respectively. Our findings are consistent with others that support a functional connection between PKD1 and PKD2 involving multiple signaling pathways that converge to induce AP-1 activity, a transcription factor that regulates different cellular programs such as proliferation, differentiation, and apoptosis. Activation of these signaling cascades may promote the full maturation of developing tubular epithelial cells, while inactivation of these signaling cascades may impair terminal differentiation and facilitate the development of renal tubular cysts.
Figures
References
-
- Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang J D, Walz G. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J Biol Chem. 1998;273:6013–6018. - PubMed
-
- Bagrodia S, Derijard B, Davis R J, Cerione R A. Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J Biol Chem. 1995;270:27995–27998. - PubMed
-
- Boyle W J, Smeal T, Defize L H, Angel P, Woodgett J R, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–584. - PubMed
-
- Chen Y, Takeshita A, Ozaki K, Kitano S, Hanazawa S. Transcriptional regulation by transforming growth factor beta of the expression of retinoic acid and retinoid X receptor genes in osteoblastic cells is mediated through AP-1. J Biol Chem. 1996;271:31602–31606. - PubMed
-
- Coso O A, Chiariello M, Yu J C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind J S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous