

Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation
- PMID: 10209028
- PMCID: PMC2133111
- DOI: 10.1083/jcb.145.2.331
Relationship between Arp2/3 complex and the barbed ends of actin filaments at the leading edge of carcinoma cells after epidermal growth factor stimulation
Abstract
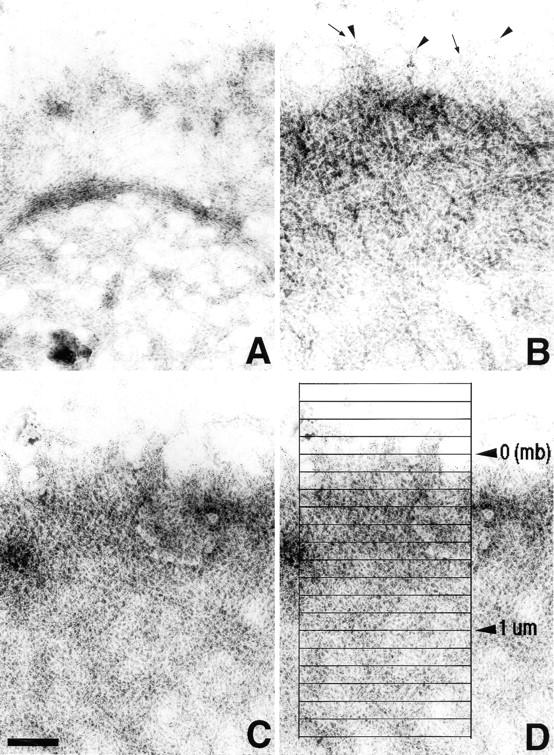
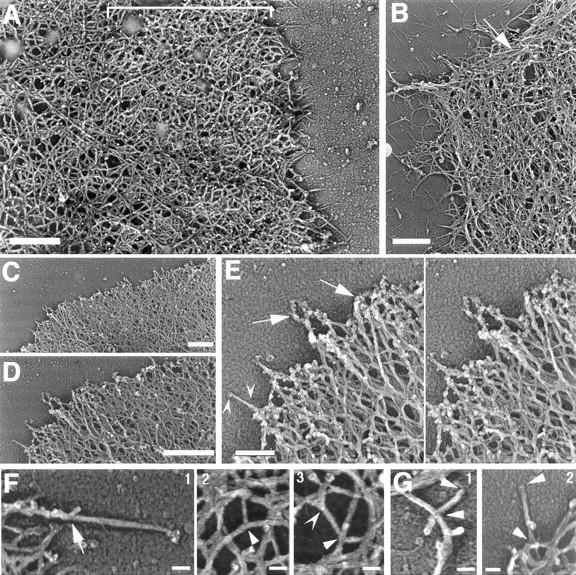
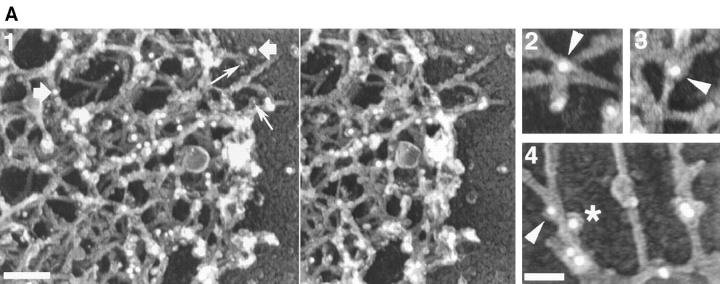
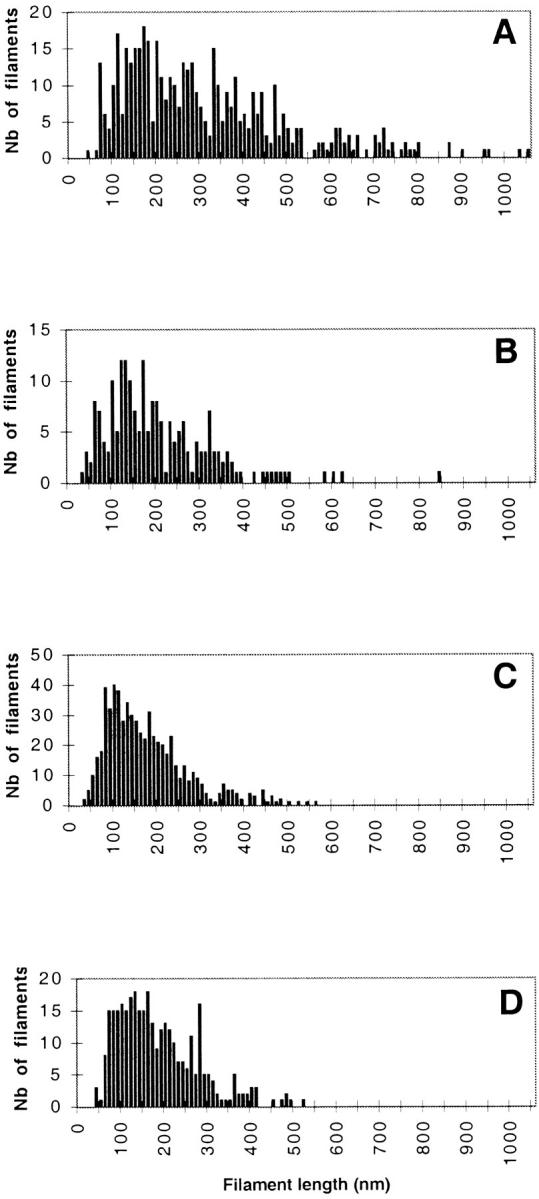
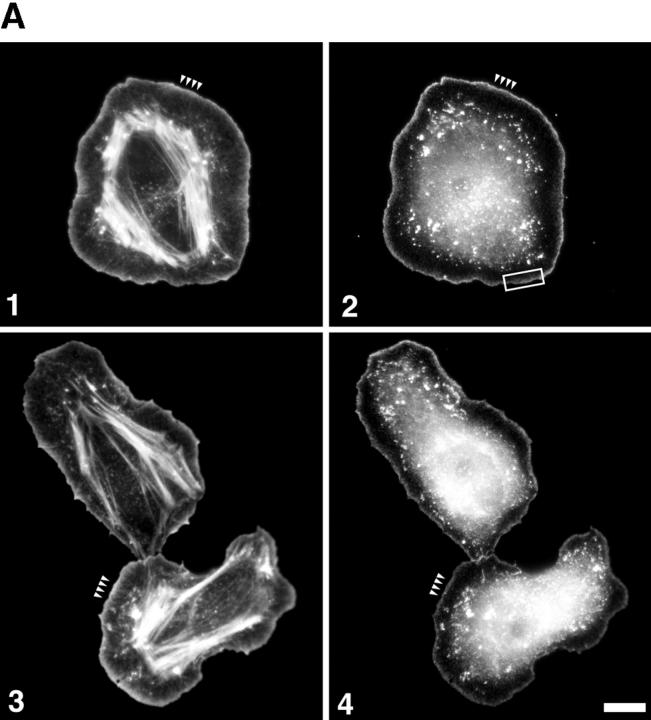
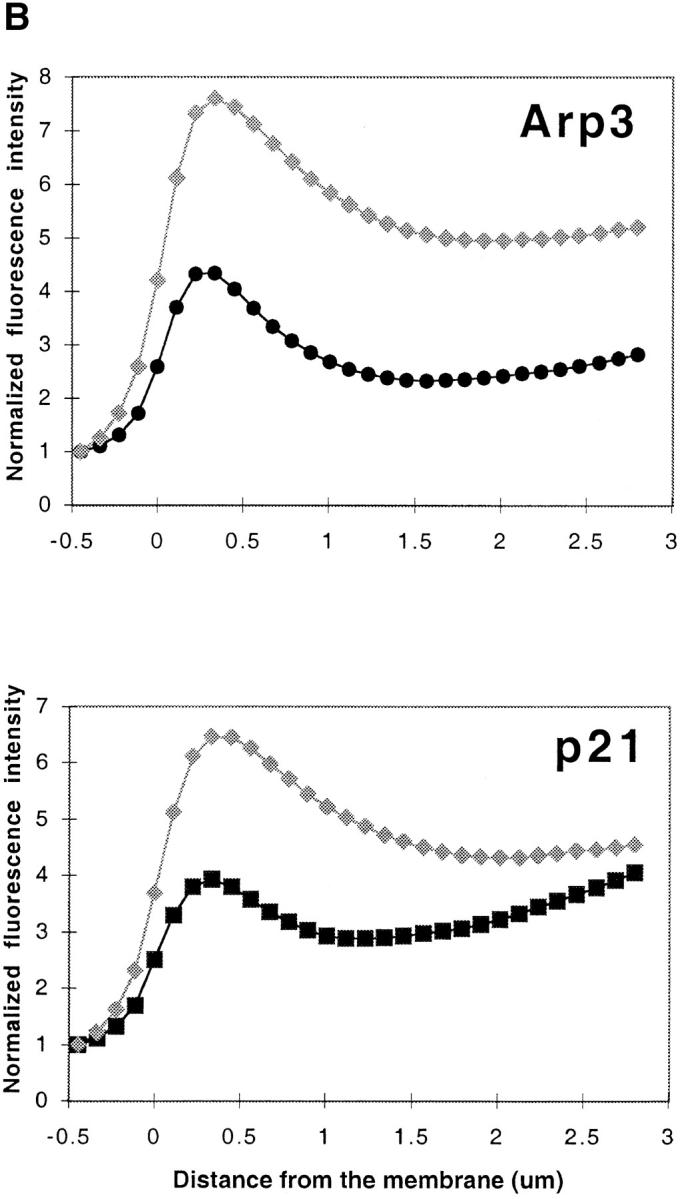
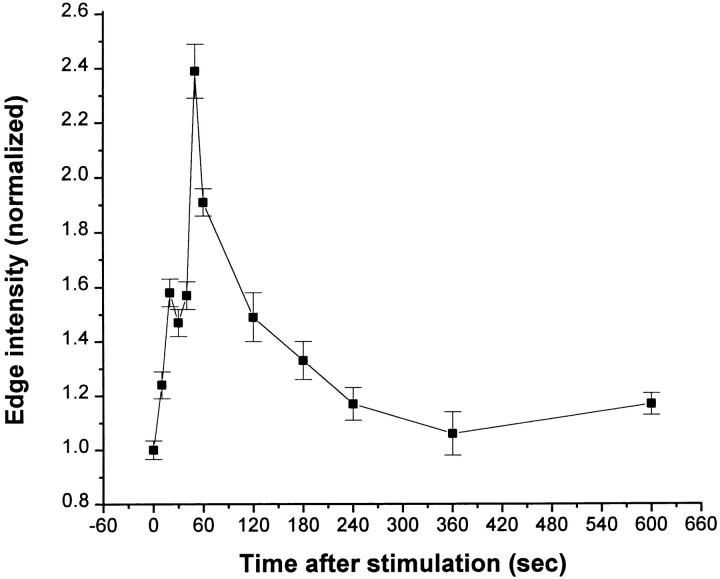
Using both light and high resolution electron microscopy, we analyzed the spatial and temporal relationships between the Arp2/3 complex and the nucleation activity that is required for lamellipod extension in mammary carcinoma cells after epidermal growth factor stimulation. A rapid two- to fourfold increase in filament barbed end number occurs transiently after stimulation and remains confined almost exclusively to the extreme outer edge of the extending lamellipod (within 100-200 nm of the plasma membrane). This is accompanied by an increase in filament density at the leading edge and a general decrease in filament length, with a specific loss of long filaments. Concomitantly, the Arp2/3 complex is recruited with a 1.5-fold increase throughout the entire cortical filament network extending 1-1.5 microm in depth from the membrane at the leading edge. The recruitment of the Arp2/3 complex at the membrane of the extending lamellipod indicates that Arp2/3 may be involved in initial generation of growing filaments. However, only a small subset of the complex present in the cortical network colocalizes near free barbed ends. This suggests that the 100-200-nm submembraneous compartment at the leading edge of the extending lamellipod constitutes a special biochemical microenvironment that favors the generation and maintenance of free barbed ends, possibly through the locally active Arp2/3 complex, severing or decreasing the on-rate of capping protein. Our results are inconsistent with the hypothesis suggesting uncapping is the dominant mechanism responsible for the generation of nucleation activity. However, they support the hypothesis of an Arp2/3-mediated capture of actin oligomers that formed close to the membrane by other mechanisms such as severing. They also support pointed-end capping by the Arp2/3 complex, accounting for its wide distribution at the leading edge.
Figures






References
-
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. I. Movements of the leading edge. Exp Cell Res. 1970;59:393–398. - PubMed
-
- Aizawa H, Fukui Y, Yahara I. Live dynamics of Dictyosteliumcofilin suggests a role in remodeling actin latticework into bundles. J Cell Sci. 1997;110:2333–2344. - PubMed
-
- Arber S, Barbayannis F, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1988;393:805–809. - PubMed
-
- Bailly M, Yan L, Whitesides GM, Condeelis JS, Segall JE. Regulation of protrusion shape and adhesion to the substratum during chemotactic responses of mammalian carcinoma cells. Exp Cell Res. 1998;241:285–299. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
