

Segmental spinal dysgenesis: neuroradiologic findings with clinical and embryologic correlation
- PMID: 10219410
- PMCID: PMC7056058
Segmental spinal dysgenesis: neuroradiologic findings with clinical and embryologic correlation
Abstract
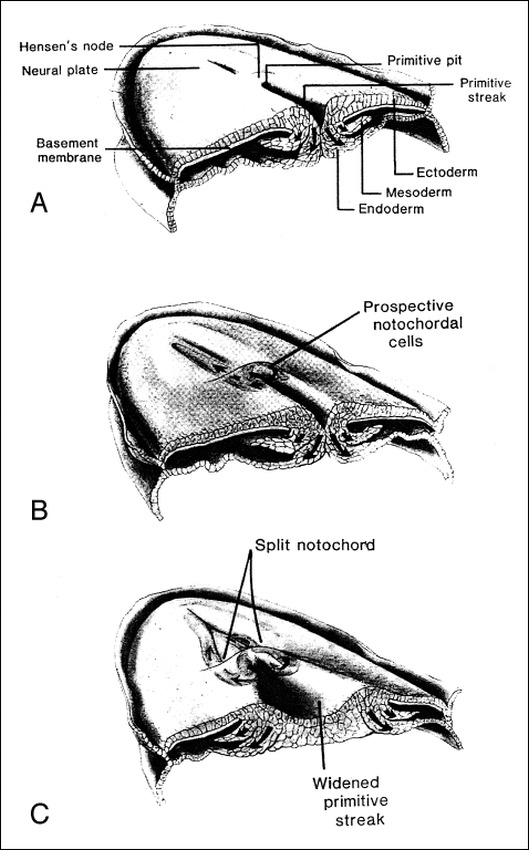
Background and purpose: Segmental spinal dysgenesis (SSD) is a rare congenital abnormality in which a segment of the spine and spinal cord fails to develop properly. Our goal was to investigate the neuroradiologic features of this condition in order to correlate our findings with the degree of residual spinal cord function, and to provide insight into the embryologic origin of this disorder. We also aimed to clarify the relationship between SSD and other entities, such as multiple vertebral segmentation defects, congenital vertebral displacement, and caudal regression syndrome (CRS).
Methods: The records of patients treated at our institutions for congenital spinal anomalies were reviewed, and 10 cases were found to satisfy the inclusion criteria for SSD. Plain radiographs were available for review in all cases. MR imaging was performed in eight patients, one of whom also underwent conventional myelography. Two other patients underwent only conventional myelography.
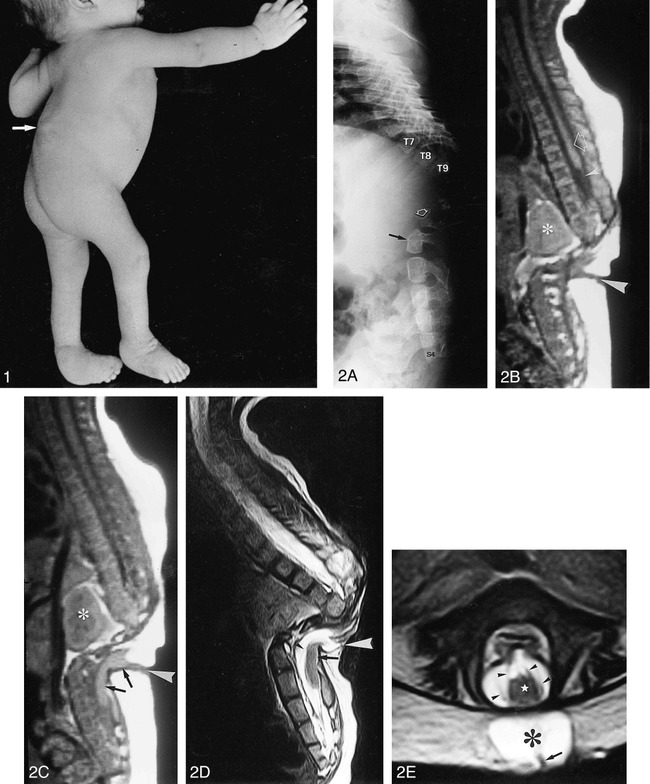
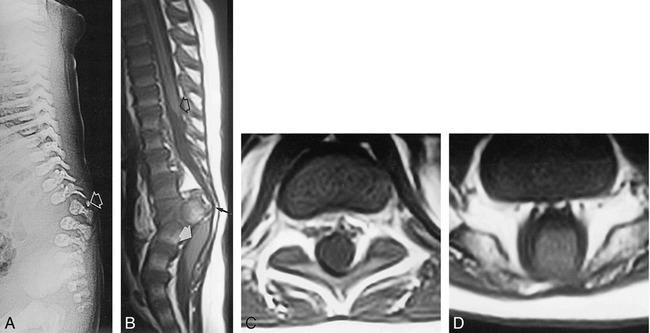
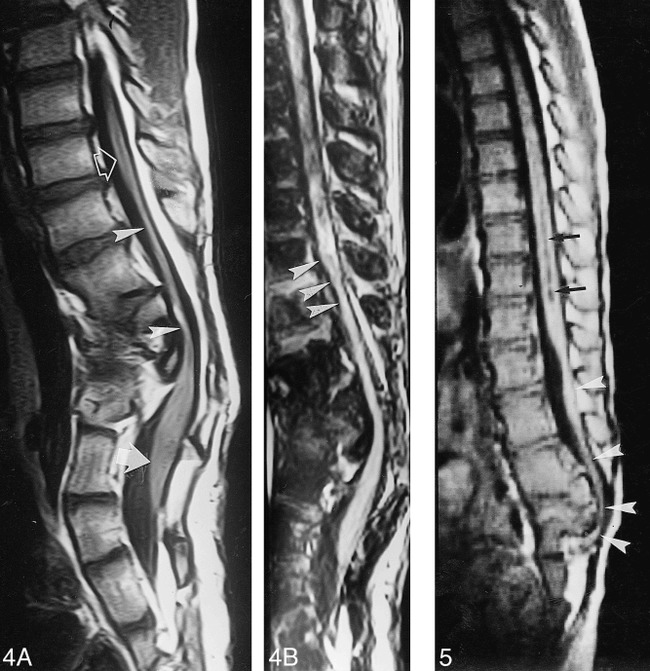
Results: Segmental vertebral anomalies involved the thoracolumbar, lumbar, or lumbosacral spine. The spinal cord at the level of the abnormality was thinned or even indiscernible, and a bulky, low-lying cord segment was present caudad to the focal abnormality in most cases. Closed spinal dysraphisms were associated in five cases, and partial sacrococcygeal agenesis in three. Renal anomalies were detected in four cases, and dextrocardia in one; all patients had a neurogenic bladder.
Conclusion: SSD is an autonomous entity with characteristic clinical and neuroradiologic features; however, SSD and CRS probably represent two faces of a single spectrum of segmental malformations of the spine and spinal cord. The neuroradiologic picture depends on the severity of the malformation and on its segmental level along the longitudinal embryonic axis. The severity of the morphologic derangement correlates with residual spinal cord function and with severity of the clinical deficit.
Figures
References
-
- Scott RM, Wolpert SM, Bartoshesky LF, Zimbler S, Karlin L. Segmental spinal dysgenesis. Neurosurgery 1988;22:739-744 - PubMed
-
- Faciszewski T, Winter RB, Lonstein JE, Sane S, Erickson D. Segmental spinal dysgenesis: a disorder different from spinal agenesis. J Bone Joint Surg [Am] 1995;77:530-537 - PubMed
-
- Fondelli MP, Cama A, Rossi A, Piatelli GL, Tortori-Donati P. Segmental spinal dysgenesis: MRI findings in 7 cases (abstr). Neuroradiology 1996;38(Suppl 2):89
-
- Raybaud CA, Couallier JF, Choux M, Binnert D. The “interrupted cord”: three cases of an unusual malformation of the spinal cord (abstr). AJNR Am J Neuroradiol 1988;9:1007
-
- McKay DW, Nason SS. Congenital duplication of the spinal canal. Spine 1980;5:390-391 - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical