

Gamma interferon production is critical for protective immunity to infection with blood-stage Plasmodium berghei XAT but neither NO production nor NK cell activation is critical
- PMID: 10225894
- PMCID: PMC115977
- DOI: 10.1128/IAI.67.5.2349-2356.1999
Gamma interferon production is critical for protective immunity to infection with blood-stage Plasmodium berghei XAT but neither NO production nor NK cell activation is critical
Abstract
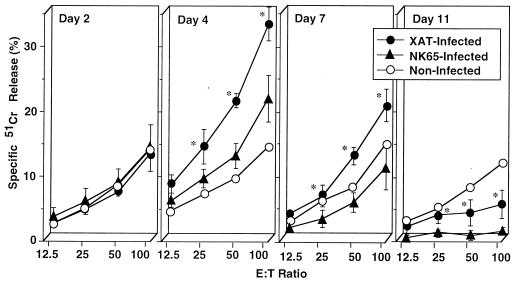
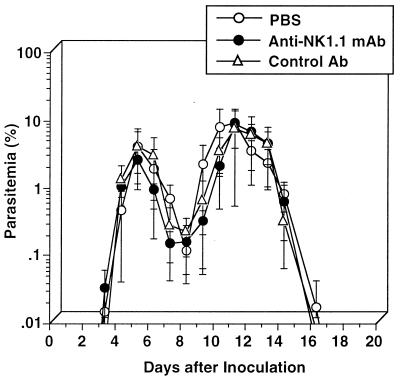
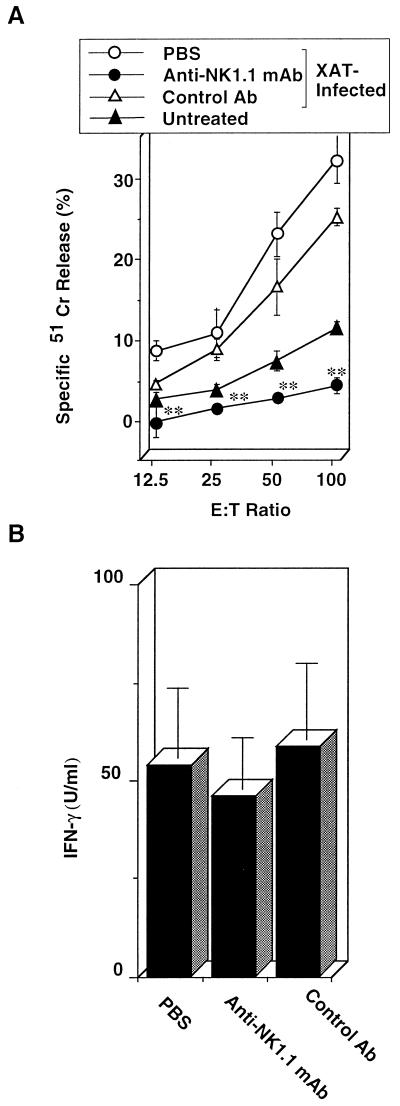
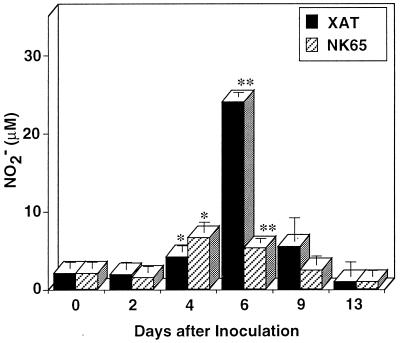
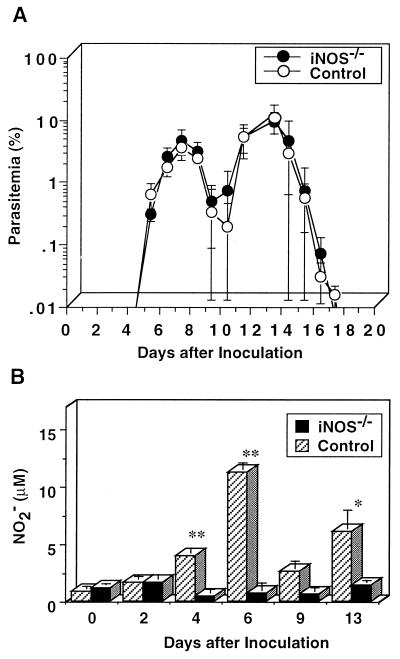
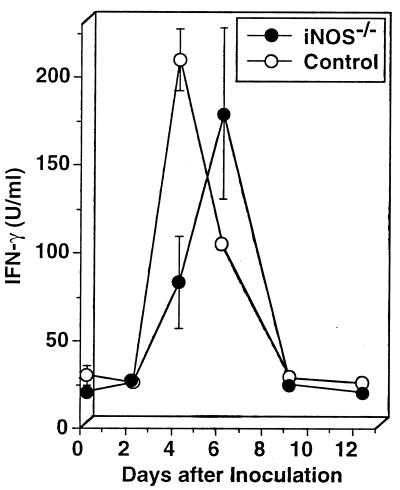
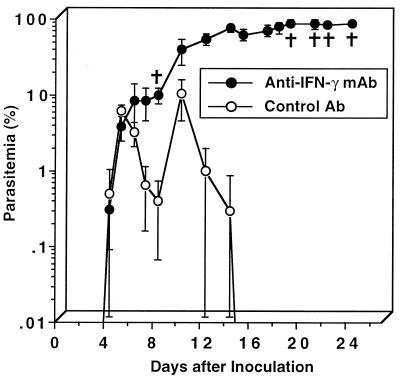
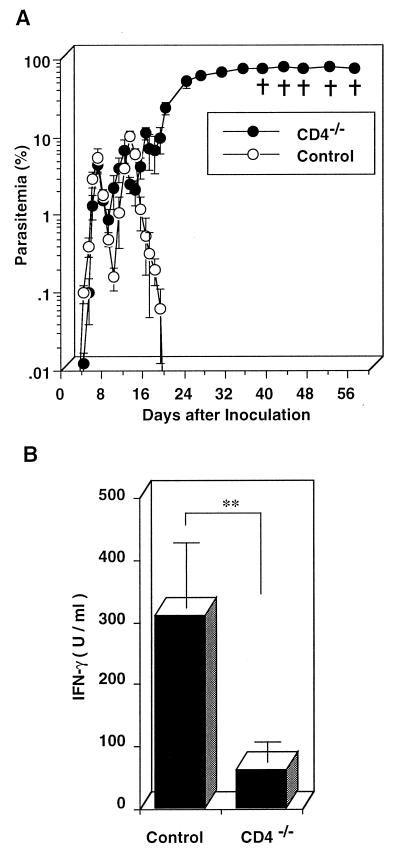
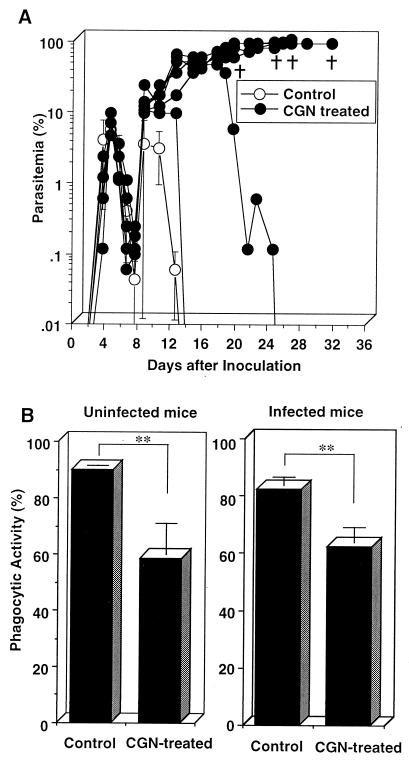
We have examined the roles of gamma interferon (IFN-gamma), nitric oxide (NO), and natural killer (NK) cells in the host resistance to infection with the blood-stage malarial parasite Plasmodium berghei XAT, an irradiation-induced attenuated variant of the lethal strain P. berghei NK65. Although the infection with P. berghei XAT enhanced NK cell lytic activity of splenocytes, depletion of NK1.1(+) cells caused by the treatment of mice with anti-NK1.1 antibody affected neither parasitemia nor IFN-gamma production by their splenocytes. The P. berghei XAT infection induced a large amount of NO production by splenocytes during the first peak of parasitemia, while P. berghei NK65 infection induced a small amount. Unexpectedly, however, mice deficient in inducible nitric oxide synthase (iNOS-/-) cleared P. berghei XAT after two peaks of parasitemia were observed, as occurred for wild-type control mice. Although the infected iNOS-/- mouse splenocytes did not produce a detectable level of NO, they produced an amount of IFN-gamma comparable to that produced by wild-type control mouse splenocytes, and treatment of these mice with neutralizing anti-IFN-gamma antibody led to the progression of parasitemia and fatal outcome. CD4(-/-) mice infected with P. berghei XAT could not clear the parasite, and all these mice died with apparently reduced IFN-gamma production. Furthermore, treatment with carrageenan increased the susceptibility of mice to P. berghei XAT infection. These results suggest that neither NO production nor NK cell activation is critical for the resistance to P. berghei XAT infection and that IFN-gamma plays an important role in the elimination of malarial parasites, possibly by the enhancement of phagocytic activity of macrophages.
Figures
References
-
- Bancroft G J. The role of natural killer cells in innate resistance to infection. Curr Opin Immunol. 1993;5:503–510. - PubMed
-
- Cherwinski H M, Schumacher J H, Brown K D, Mosmann T R. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J Exp Med. 1987;166:1229–1244. - PMC - PubMed
-
- Clark I A, Butcher G A, Buffinton G D, Hunt N H, Cowden W B. Toxicity of certain products of lipid peroxidation to the human malaria parasite Plasmodium falciparum. Biochem Pharmacol. 1987;36:543–546. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
