

Murine acid alpha-glucosidase: cell-specific mRNA differential expression during development and maturation
- PMID: 10233847
- PMCID: PMC1866561
- DOI: 10.1016/s0002-9440(10)65361-8
Murine acid alpha-glucosidase: cell-specific mRNA differential expression during development and maturation
Abstract
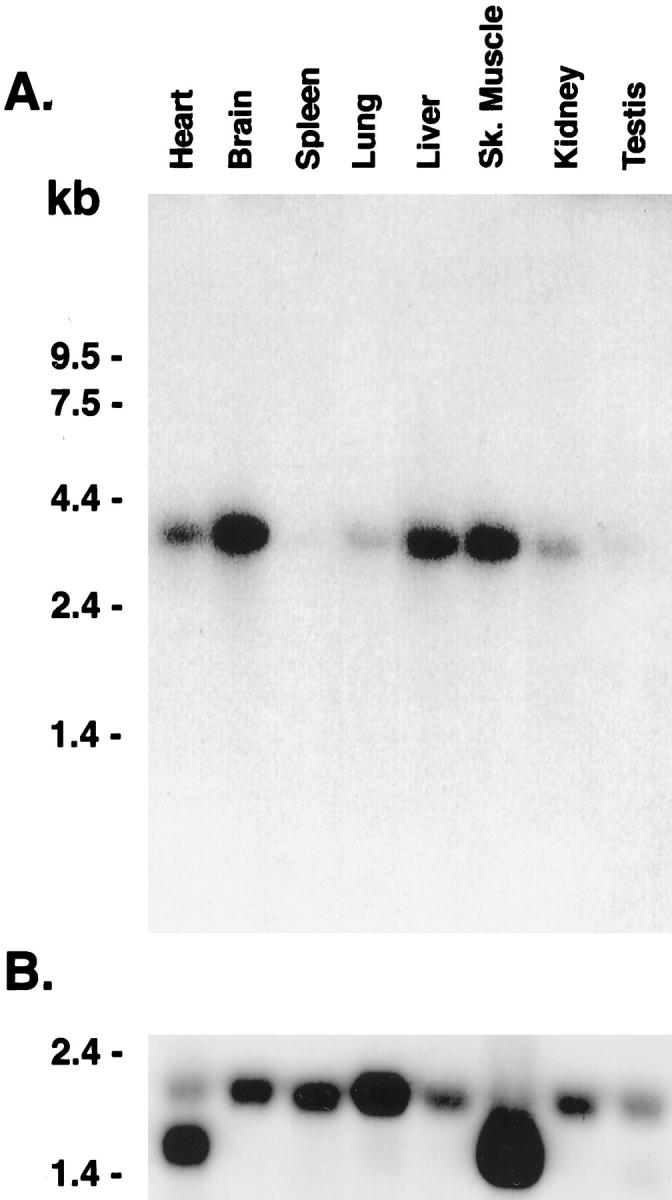
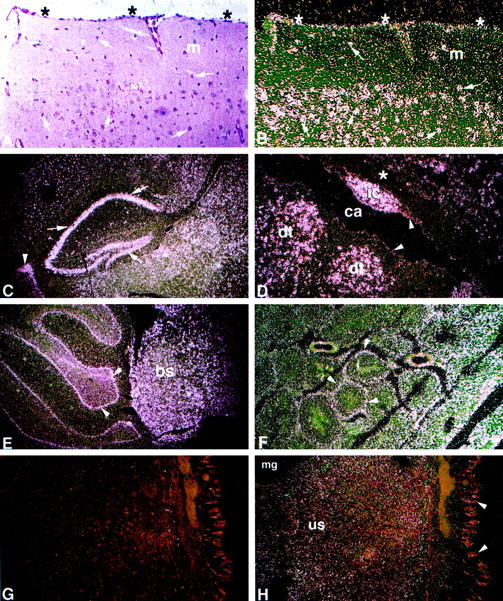
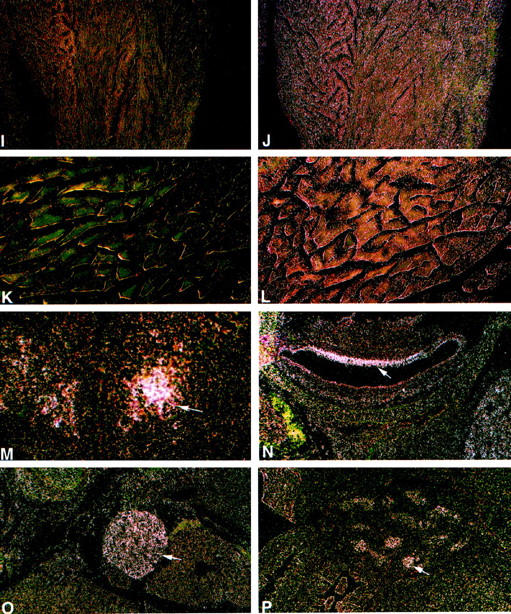
Acid alpha-glucosidase (GAA) cleaves the alpha1-4 and alpha1-6 glycosidic linkages of glycogen and related alpha-glucosyl substrates within lysosomes. Its deficiency results in glycogen storage disease type II (GSDII) variants including Pompe disease. To gain insight into the tissue patterns of involvement by glycogen storage in GSDII, GAA mRNA expression in mouse tissues was evaluated by Northern blot and in situ hybridization analyses. Extensive temporal and spatial variation of GAA mRNA was observed. During preterm maturation, GAA mRNA levels of whole mice progressively increased as assessed by Northern analysis. By in situ hybridization with GAA antisense mRNA, low signals were detected in most tissues throughout gestation. However, increased expression in specific cell types of different tissues was observed beginning at 16 days post coitum in developing brain neurons, primitive inner ear cells, and seminiferous tubular epithelium. In adult mice, whole-organ GAA mRNA levels were highest in brain, moderate in heart, liver, and skeletal muscle, and lowest in the series kidney > lung > testis > spleen. By in situ hybridization, the highest-intensity signals were in neurons of the central and peripheral nervous systems whereas neuroglial cells had only low-level signal. Signals of moderate intensity were in cardiomyocytes whereas low signals were in hepatocytes and skeletal muscle myocytes and very low in cells of the lungs, thymus, pancreas, spleen, and adrenal glands. However, testicular Sertoli cells and kidney tubular epithelial cells had significant signals even though surrounding cells had very low signals. The discrete temporal and spatial variations of GAA mRNA during development indicate different physiological roles for this enzyme in various cell types and developmental stages.
Figures
References
-
- Hers HG: α-Glucosidase in generalized glycogen-storage disease (Pompe’s disease). Biochem J 1963, 86:11-16 - PubMed
-
- Brown BI, Brown DH: The subcellular distribution of enzymes in type II glycogenosis and the occurrence of oligo α-1,4-glucan glucohydrolase in human tissues. Biochim Biophys Acta 1981, 110:124-133 - PubMed
-
- Hirschhorn R: Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. Scriver CR Beaudet AL Sly WL Valle D eds. The Metabolic and Molecular Bases of Inherited Disease. 1995, :pp 2443-2464 McGraw Hill, New York
-
- Mancall EL, Aponte GE, Berry RG: Pompe’s disease (diffuse glycogenosis) with neuronal storage. J Neuropathol Exp Neurol 1965, 24:85-96 - PubMed
-
- Hug G, Schubert WK: Glycogenosis type II. Arch Pathol 1967, 84:141-152 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
