

Evolution of neoplastic cell lineages in Barrett oesophagus
- PMID: 10319873
- PMCID: PMC1559997
- DOI: 10.1038/8816
Evolution of neoplastic cell lineages in Barrett oesophagus
Abstract
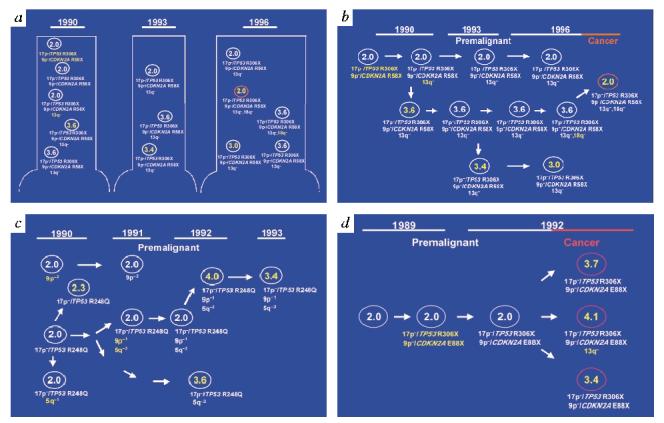
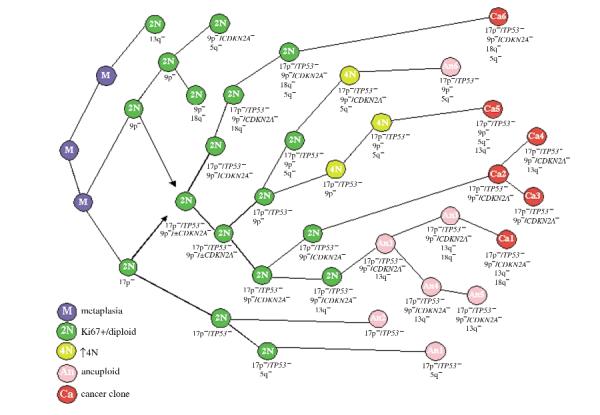
It has been hypothesized that neoplastic progression develops as a consequence of an acquired genetic instability and the subsequent evolution of clonal populations with accumulated genetic errors. Accordingly, human cancers and some premalignant lesions contain multiple genetic abnormalities not present in the normal tissues from which the neoplasms arose. Barrett oesophagus (BE) is a premalignant condition which predisposes to oesophageal adenocarcinoma (EA) that can be biopsied prospectively over time because endoscopic surveillance is recommended for early detection of cancer. In addition, oesophagectomy specimens frequently contain the premalignant epithelium from which the cancer arose. Neoplastic progression in BE is associated with alterations in TP53 (also known as p53) and CDKN2A (also known as p16) and non-random losses of heterozygosity (LOH). Aneuploid or increased 4N populations occur in more than 90-95% of EAs, arise in premalignant epithelium and predict progression. We have previously shown in small numbers of patients that disruption of TP53 and CDKN2A typically occurs before aneuploidy and cancer. Here, we determine the evolutionary relationships of non-random LOH, TP53 and CDKN2A mutations, CDKN2A CpG-island methylation and ploidy during neoplastic progression. Diploid cell progenitors with somatic genetic or epigenetic abnormalities in TP53 and CDKN2A were capable of clonal expansion, spreading to large regions of oesophageal mucosa. The subsequent evolution of neoplastic progeny frequently involved bifurcations and LOH at 5q, 13q and 18q that occurred in no obligate order relative to each other, DNA-content aneuploidy or cancer. Our results indicate that clonal evolution is more complex than predicted by linear models.
Figures
References
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. - PubMed
-
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. - PubMed
-
- Boland CR, Sato J, Appelman HD, Besalier RS, Feinberg AP. Microallelotyping defines the sequence and tempo of allelic losses at tumor suppressor gene loci during colorectal cancer progression. Nature Med. 1995;1:902–909. - PubMed
-
- Spechler SJ. Endoscopic surveillance for patients with Barrett esophagus: does the cancer risk justify the practice. Ann. Intern. Med. 1987;106:902–904. - PubMed
-
- Levine DS, et al. An endoscopic biopsy protocol can differentiate high-grade dysplasia from early adenocarcinoma in Barrett’s esophagus. Gastroenterology. 1993;105:40–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
