

Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders
- PMID: 10329600
- PMCID: PMC1866602
- DOI: 10.1016/S0002-9440(10)65401-6
Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders
Abstract
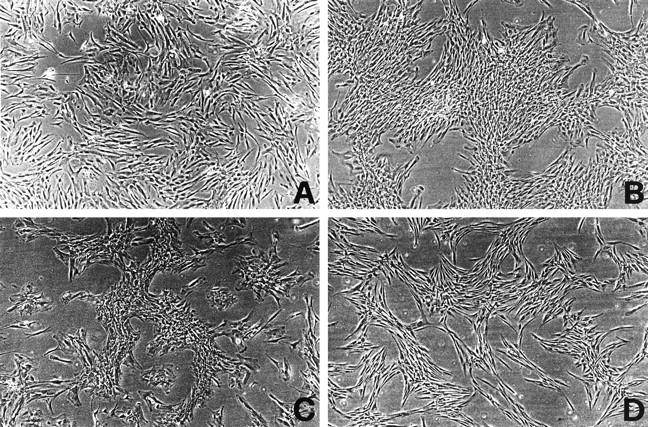
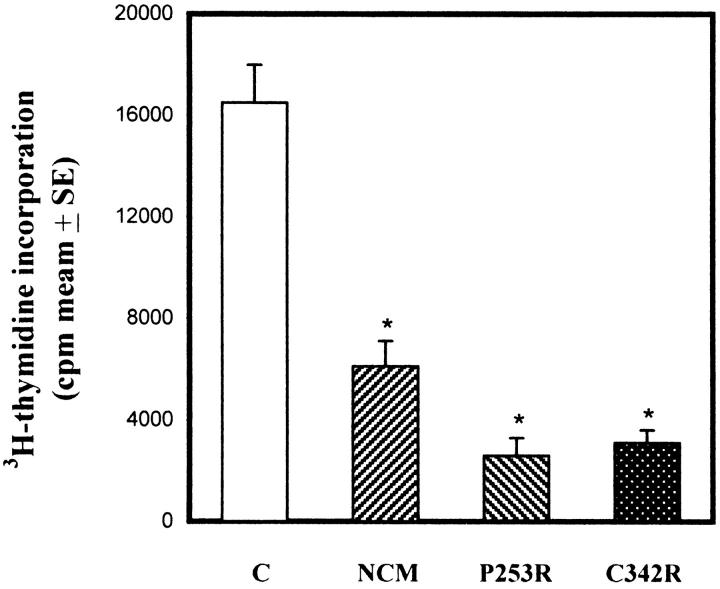
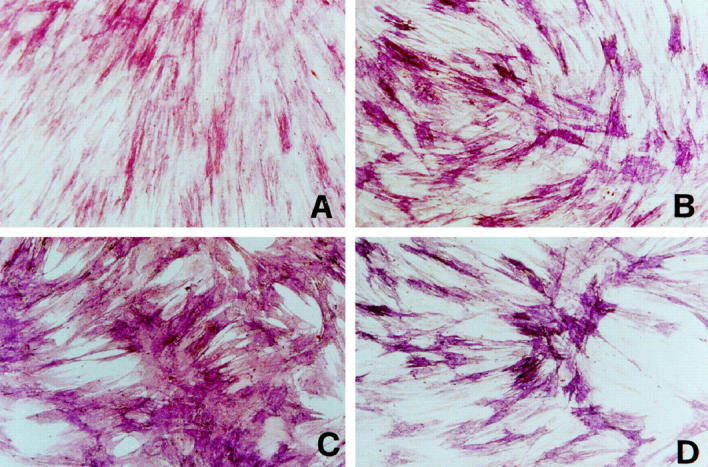
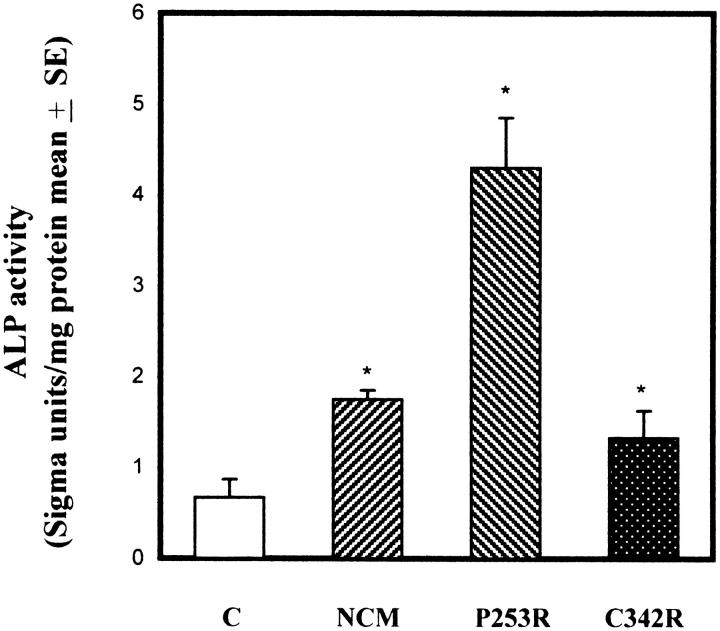
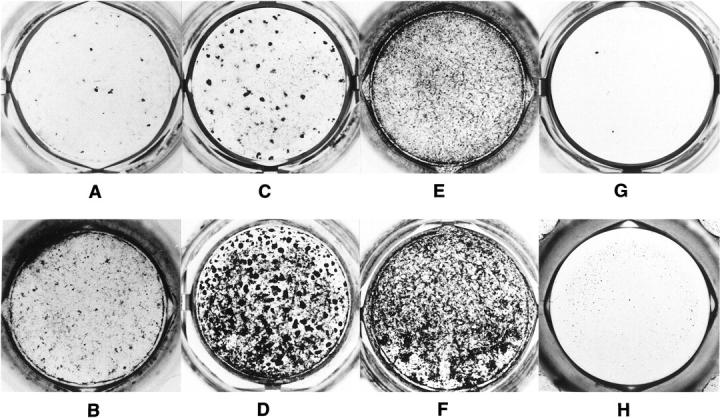
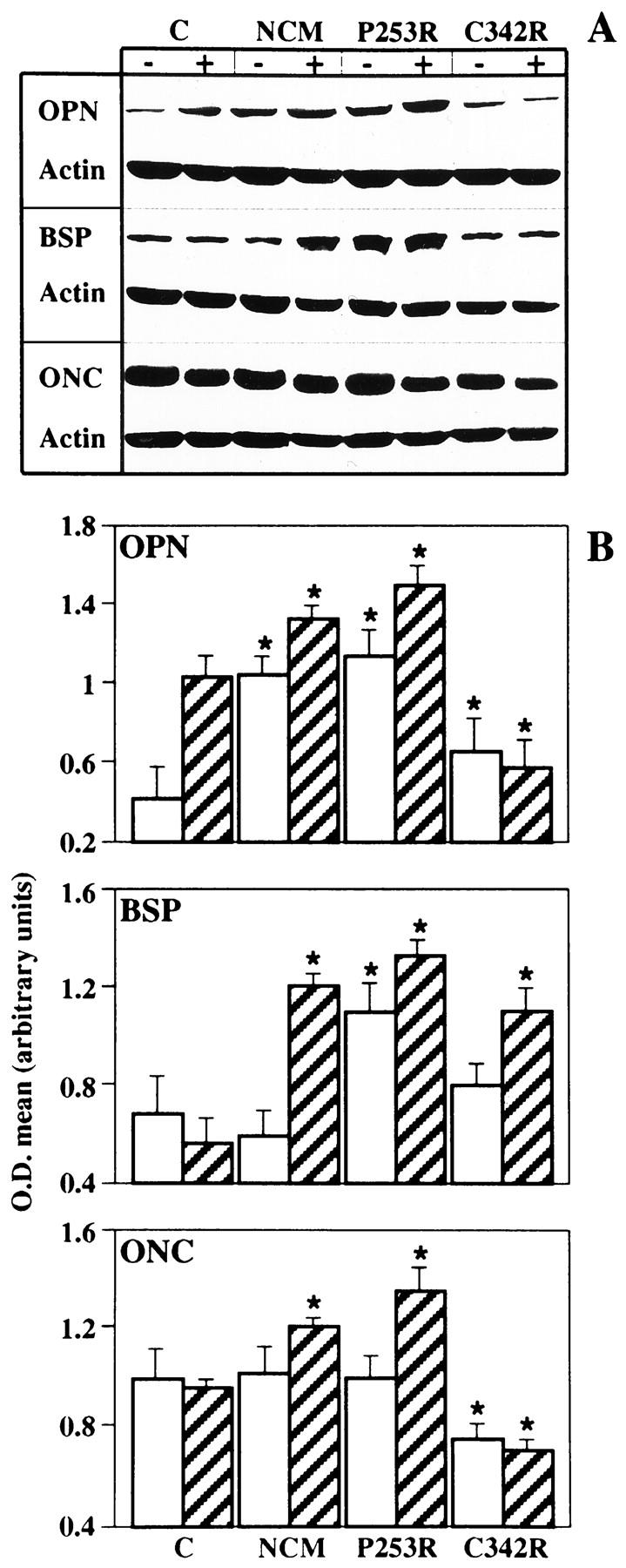
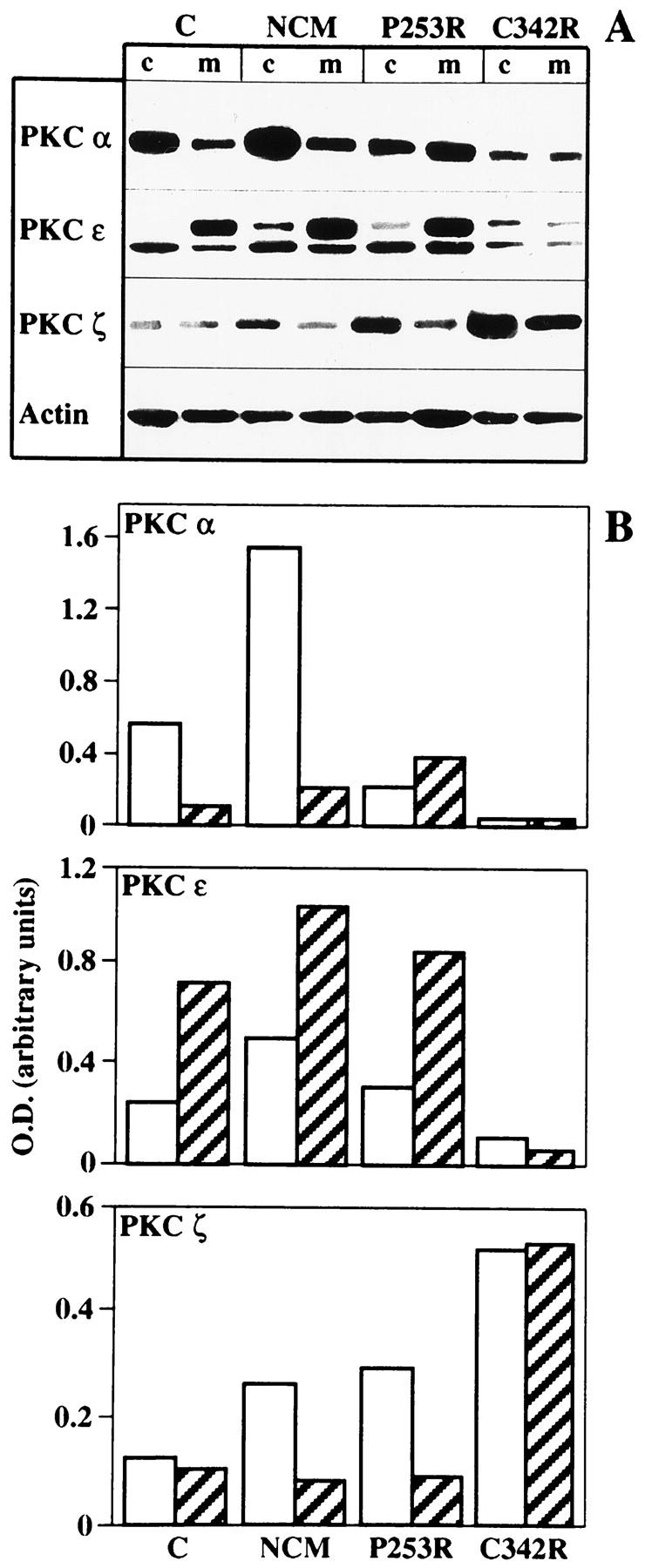
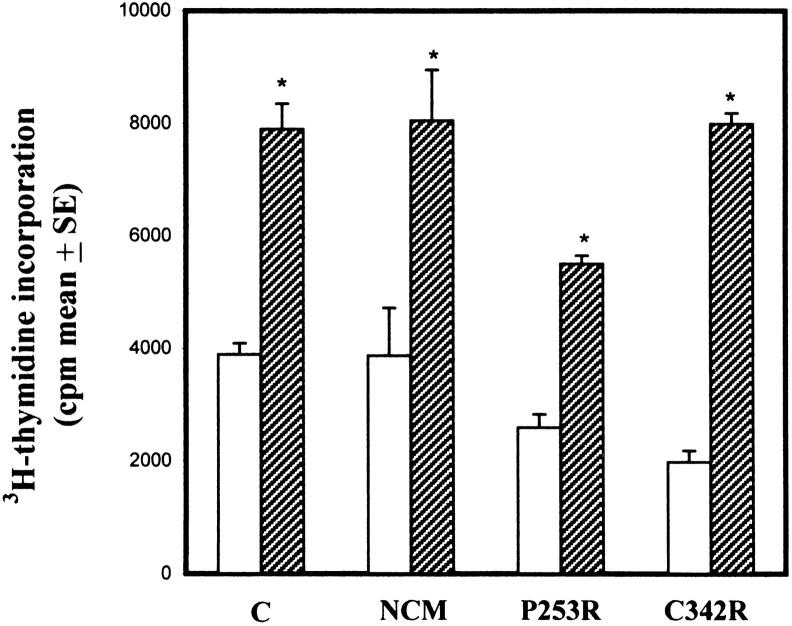
Craniosynostoses are a heterogeneous group of disorders characterized by premature fusion of cranial sutures. Mutations in fibroblast growth factor receptors (FGFRs) have been associated with a number of such conditions. Nevertheless, the cellular mechanism(s) involved remain unknown. We analyzed cell proliferation and differentiation in osteoblasts obtained from patients with three genetically and clinically distinct craniosynostoses: Pfeiffer syndrome carrying the FGFR2 C342R substitution, Apert syndrome with FGFR2 P253R change, and a nonsyndromic craniosynostosis without FGFR canonic mutations, as compared with control osteoblasts. Osteoblasts from craniosynostotic patients exhibited a lower proliferation rate than control osteoblasts. P253R and nonsyndromic craniosynostosis osteoblasts showed a marked differentiated phenotype, characterized by high alkaline phosphatase activity, increased mineralization and expression of noncollagenous matrix proteins, associated with high expression and activation of protein kinase Calpha and protein kinase Cepsilon isoenzymes. By contrast, the low proliferation rate of C342R osteoblasts was not associated with a differentiated phenotype. Although they showed higher alkaline phosphatase activity than control, C342R osteoblasts failed to mineralize and expressed low levels of osteopontin and osteonectin and high protein kinase Czeta levels. Stimulation of proliferation and inhibition of differentiation were observed in all cultures on FGF2 treatment. Our results suggest that an anticipated proliferative/differentiative switch, associated with alterations of the FGFR transduction pathways, could be the causative common feature in craniosynostosis and that mutations in distinct FGFR2 domains are associated with an in vitro heterogeneous differentiative phenotype.
Figures
References
-
- Cohen MM, Jr: Craniosynostosis: diagnosis, evaluation and management. 1986. Raven Press, New York
-
- Di Rocco C, Velardi F: Surgical management of craniosynostosis. Galli G eds. Craniosynostosis. 1984, :pp 181-248 CRC Press, Boca Raton, FL
-
- McKusick VA: Mendelian inheritance in man. Catalogs of Human Genes and Genetic Disorders. 1994, Johns Hopkins University Press, Baltimore
-
- Bonaventure J, Rousseau F, Legeai-Mallet L, Le Merrer M, Munnich A, Maroteaux P: Common mutations in the fibroblast growth factor receptor 3 (FGFR3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am J Med Genet 1996, 63:148-154 - PubMed
-
- Wilkie AOM: Genes and mechanisms. Hum Mol Genet 1997, 6:1647-1656 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
