

A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5
- PMID: 10330170
- PMCID: PMC104389
- DOI: 10.1128/MCB.19.6.4289
A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5
Abstract
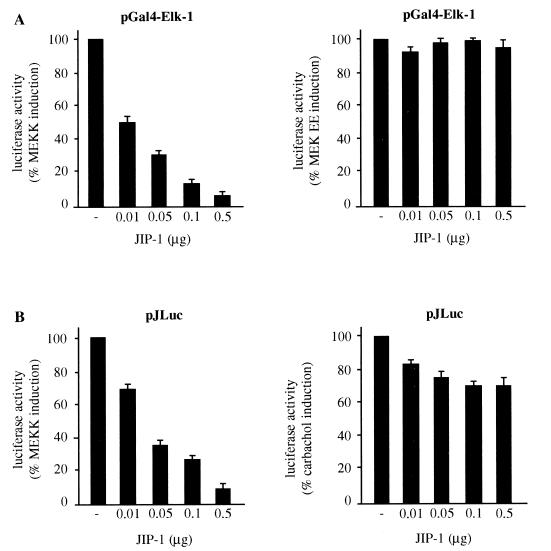
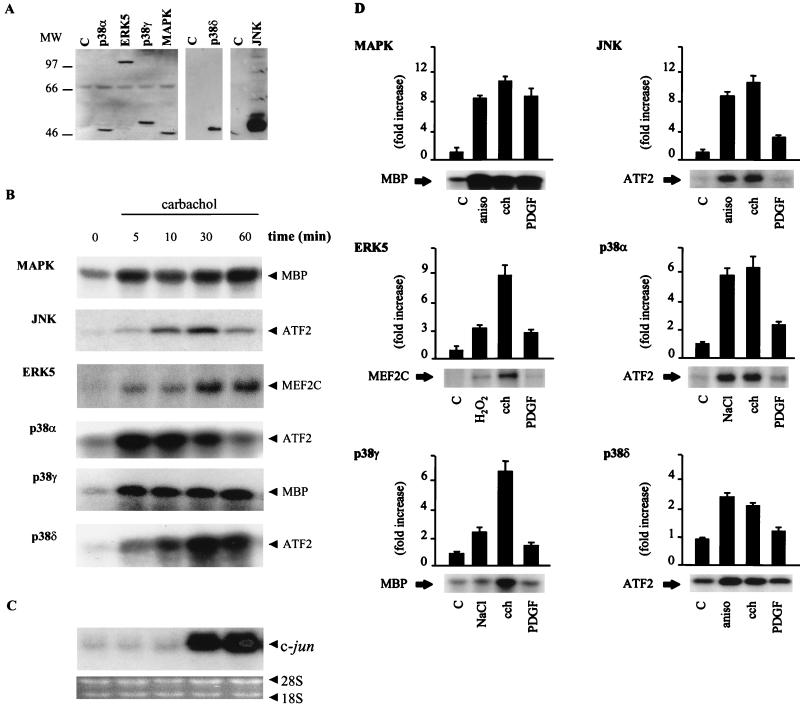
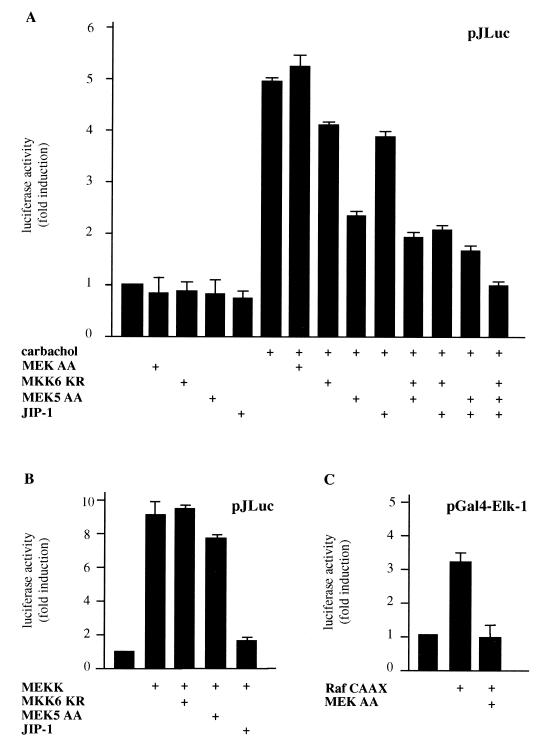
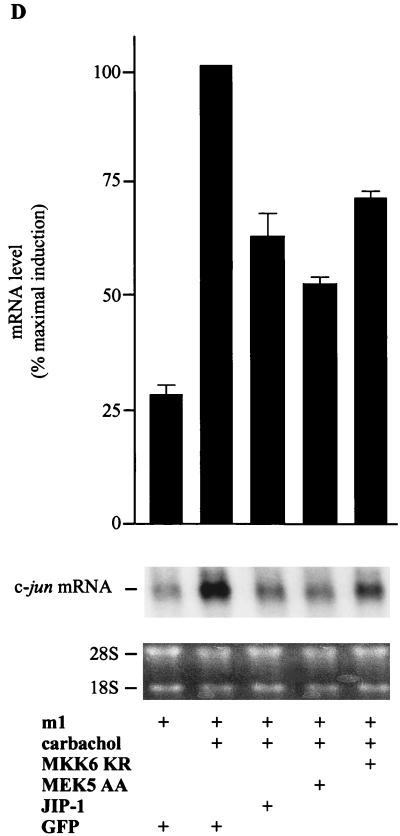
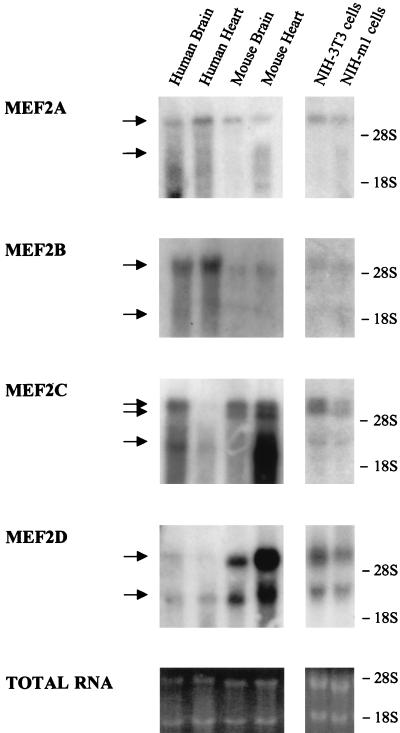
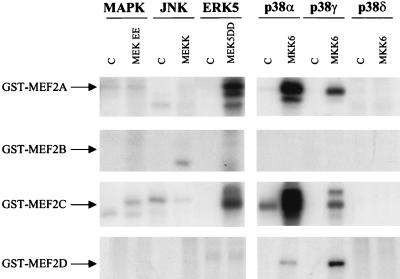
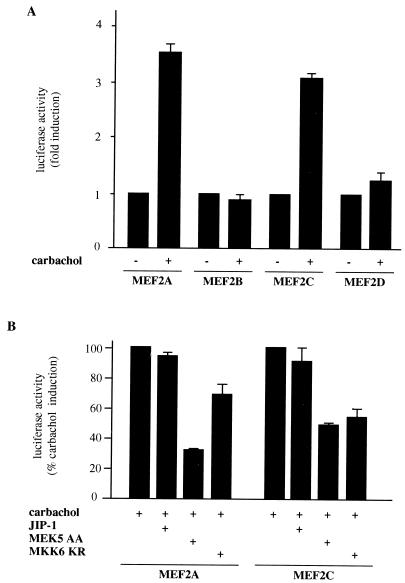
The expression of the c-jun proto-oncogene is rapidly induced in response to mitogens acting on a large variety of cell surface receptors. The resulting functional activity of c-Jun proteins appears to be critical for cell proliferation. Recently, we have shown that a large family of G protein-coupled receptors (GPCRs), represented by the m1 muscarinic receptor, can initiate intracellular signaling cascades that result in the activation of mitogen-activated protein kinases (MAPK) and c-Jun NH2-terminal kinases (JNK) and that the activation of JNK but not of MAPK correlated with a remarkable increase in the expression of c-jun mRNA. Subsequently, however, we obtained evidence that GPCRs can potently stimulate the activity of the c-jun promoter through MEF2 transcription factors, which do not act downstream from JNK. In view of these observations, we set out to investigate further the nature of the signaling pathway linking GPCRs to the c-jun promoter. Utilizing NIH 3T3 cells, we found that GPCRs can activate the c-jun promoter in a JNK-independent manner. Additionally, we demonstrated that these GPCRs can elevate the activity of novel members of the MAPK family, including ERK5, p38alpha, p38gamma, and p38delta, and that the activation of certain kinases acting downstream from MEK5 (ERK5) and MKK6 (p38alpha and p38gamma) is necessary to fully activate the c-jun promoter. Moreover, in addition to JNK, ERK5, p38alpha, and p38gamma were found to stimulate the c-jun promoter by acting on distinct responsive elements. Taken together, these results suggest that the pathway linking GPCRs to the c-jun promoter involves the integration of numerous signals transduced by a highly complex network of MAPK, rather than resulting from the stimulation of a single linear protein kinase cascade. Furthermore, our findings suggest that each signaling pathway affects one or more regulatory elements on the c-jun promoter and that the transcriptional response most likely results from the temporal integration of each of these biochemical routes.
Figures




References
-
- Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. - PubMed
-
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. - PubMed
-
- Ball A R, Jr, Bos T J, Loliger C, Nagata L P, Nishimura T, Su H, Tsuchie H, Vogt P K. Jun: oncogene and transcriptional regulator. Cold Spring Harbor Symp Quant Biol. 1988;53:687–693. - PubMed
-
- Benbrook D M, Jones N C. Heterodimer formation between CREB and JUN proteins. Oncogene. 1990;5:295–302. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous