

Role of region C in regulation of the heat shock gene-specific sigma factor of Escherichia coli, sigma32
- PMID: 10348869
- PMCID: PMC93824
- DOI: 10.1128/JB.181.11.3552-3561.1999
Role of region C in regulation of the heat shock gene-specific sigma factor of Escherichia coli, sigma32
Abstract
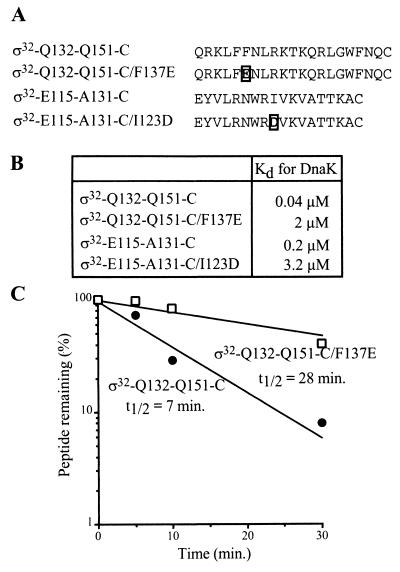
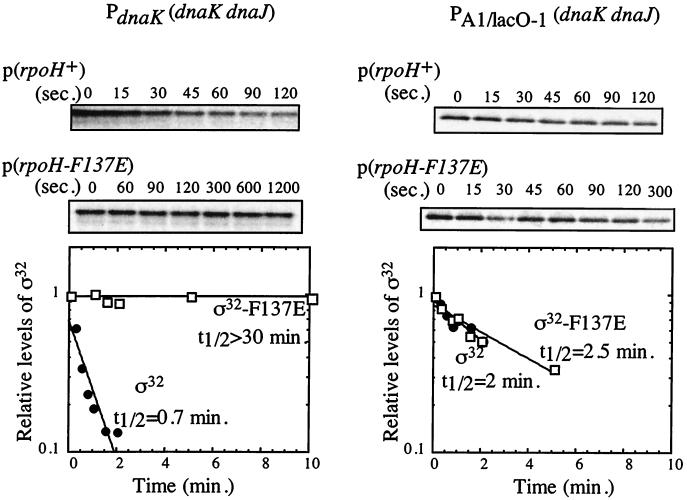
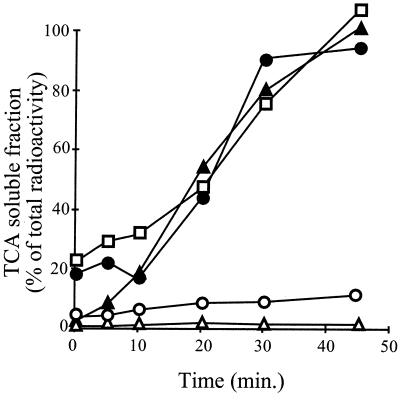
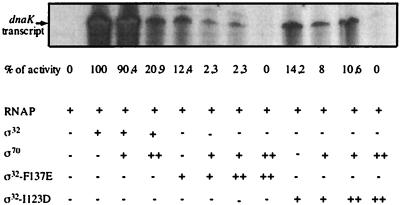
Expression of heat shock genes is controlled in Escherichia coli by the antagonistic action of the sigma32 subunit of RNA polymerase and the DnaK chaperone system, which inactivates sigma32 by stress-dependent association and mediates sigma32 degradation by the FtsH protease. A stretch of 23 residues (R122 to Q144) conserved among sigma32 homologs, termed region C, was proposed to play a role in sigma32 degradation, and peptide analysis identified two potential DnaK binding sites central and peripheral to region C. Region C is thus a prime candidate for mediating stress control of sigma32, a hypothesis that we tested in the present study. A peptide comprising the central DnaK binding site was an excellent substrate for FtsH, while a peptide comprising the peripheral DnaK binding site was a poor substrate. Replacement of a single hydrophobic residue in each DnaK binding site by negatively charged residues (I123D and F137E) strongly decreased the binding of the peptides to DnaK and the degradation by FtsH. However, introduction of these and additional region C alterations into the sigma32 protein did not affect sigma32 degradation in vivo and in vitro or DnaK binding in vitro. These findings do not support a role for region C in sigma32 control by DnaK and FtsH. Instead, the sigma32 mutants had reduced affinities for RNA polymerase and decreased transcriptional activities in vitro and in vivo. Furthermore, cysteines inserted into region C allowed cysteine-specific cross-linking of sigma32 to RNA polymerase. Region C thus confers on sigma32 a competitive advantage over other sigma factors to bind RNA polymerase and thereby contributes to the rapidity of the heat shock response.
Figures




Similar articles
-
Regulatory region C of the E. coli heat shock transcription factor, sigma32, constitutes a DnaK binding site and is conserved among eubacteria.J Mol Biol. 1996 Mar 15;256(5):829-37. doi: 10.1006/jmbi.1996.0129. J Mol Biol. 1996. PMID: 8601834
-
Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli.Mol Microbiol. 1998 Nov;30(3):567-81. doi: 10.1046/j.1365-2958.1998.01090.x. Mol Microbiol. 1998. PMID: 9822822
-
A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32.EMBO J. 1996 Feb 1;15(3):607-17. EMBO J. 1996. PMID: 8599944 Free PMC article.
-
The heat shock response of Escherichia coli.Int J Food Microbiol. 2000 Apr 10;55(1-3):3-9. doi: 10.1016/s0168-1605(00)00206-3. Int J Food Microbiol. 2000. PMID: 10791710 Review.
-
Revival of the Escherichia coli heat shock response after two decades with a small Hsp in a critical but distinct act.Biol Chem. 2025 Jan 7;406(1-2):29-33. doi: 10.1515/hsz-2024-0140. Print 2025 Jan 29. Biol Chem. 2025. PMID: 39760265 Review.
Cited by
-
Structural Insight into the Mechanism of σ32-Mediated Transcription Initiation of Bacterial RNA Polymerase.Biomolecules. 2023 Apr 25;13(5):738. doi: 10.3390/biom13050738. Biomolecules. 2023. PMID: 37238608 Free PMC article.
-
Activity of Rhodobacter sphaeroides RpoHII, a second member of the heat shock sigma factor family.J Bacteriol. 2006 Aug;188(16):5712-21. doi: 10.1128/JB.00405-06. J Bacteriol. 2006. PMID: 16885439 Free PMC article.
-
Structure-function studies of Escherichia coli RpoH (sigma32) by in vitro linker insertion mutagenesis.J Bacteriol. 2003 May;185(9):2731-8. doi: 10.1128/JB.185.9.2731-2738.2003. J Bacteriol. 2003. PMID: 12700252 Free PMC article.
-
Convergence of the transcriptional responses to heat shock and singlet oxygen stresses.PLoS Genet. 2012 Sep;8(9):e1002929. doi: 10.1371/journal.pgen.1002929. Epub 2012 Sep 13. PLoS Genet. 2012. PMID: 23028346 Free PMC article.
-
The C terminus of sigma(32) is not essential for degradation by FtsH.J Bacteriol. 2001 Oct;183(20):5911-7. doi: 10.1128/JB.183.20.5911-5917.2001. J Bacteriol. 2001. PMID: 11566990 Free PMC article.
References
-
- Blaszczak A, Georgopoulos C, Liberek K. On the mechanism of FtsH-dependent degradation of the ς32 transcriptional regulator of Escherichia coli and the role of the DnaK chaperone machine. Mol Microbiol. 1999;31:157–166. - PubMed
-
- Bukau B. Regulation of the E. coli heat shock response. Mol Microbiol. 1993;9:671–680. - PubMed
-
- Bukau B, editor. Molecular chaperones and folding catalysts—regulation, cellular function and mechanisms. Amsterdam, The Netherlands: Harwood Academic Publishers; 1999.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
