

Identification of Grb2 as a novel binding partner of tumor necrosis factor (TNF) receptor I
- PMID: 10359574
- PMCID: PMC2193078
- DOI: 10.1084/jem.189.11.1707
Identification of Grb2 as a novel binding partner of tumor necrosis factor (TNF) receptor I
Abstract
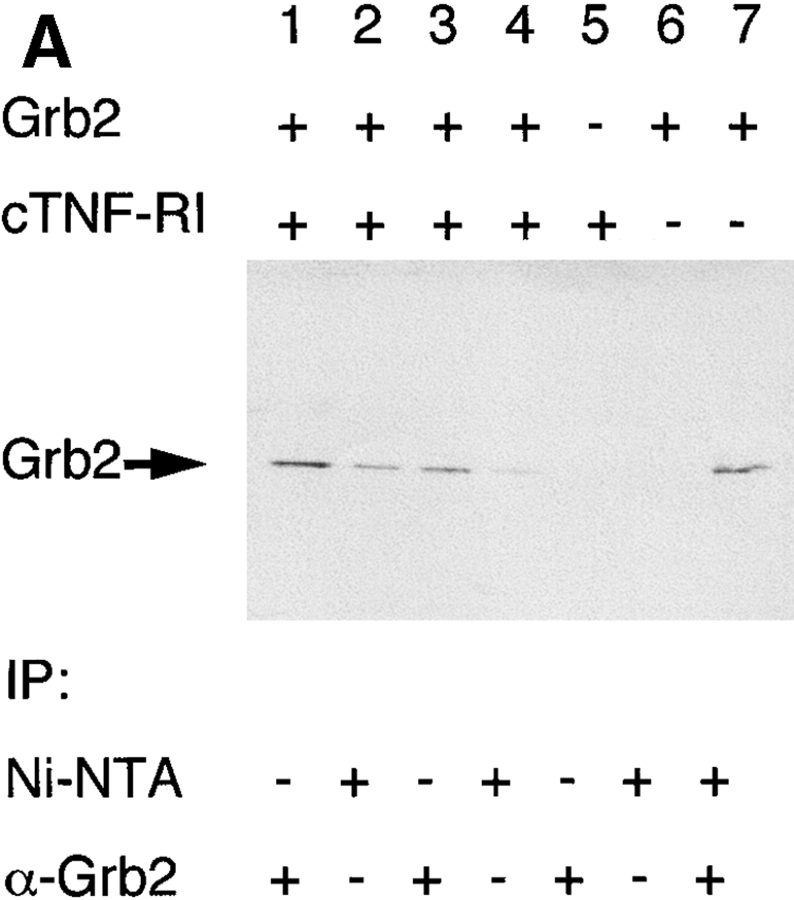
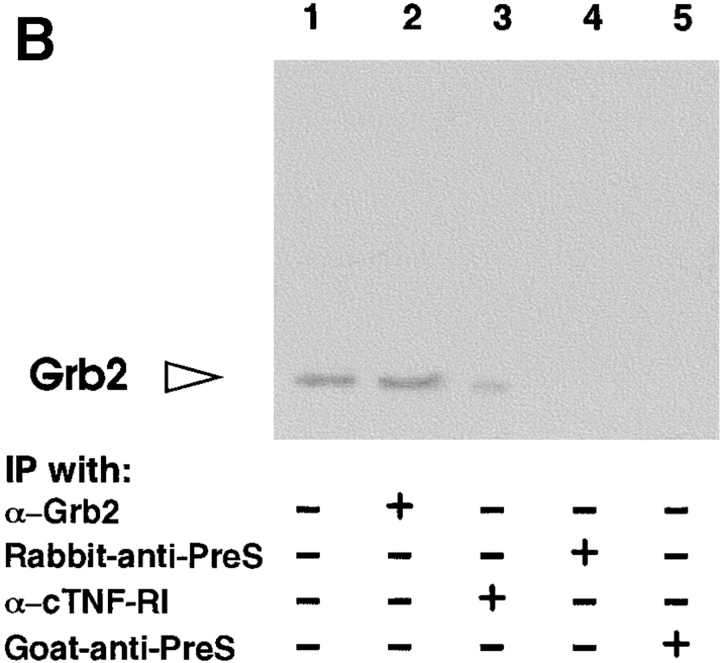
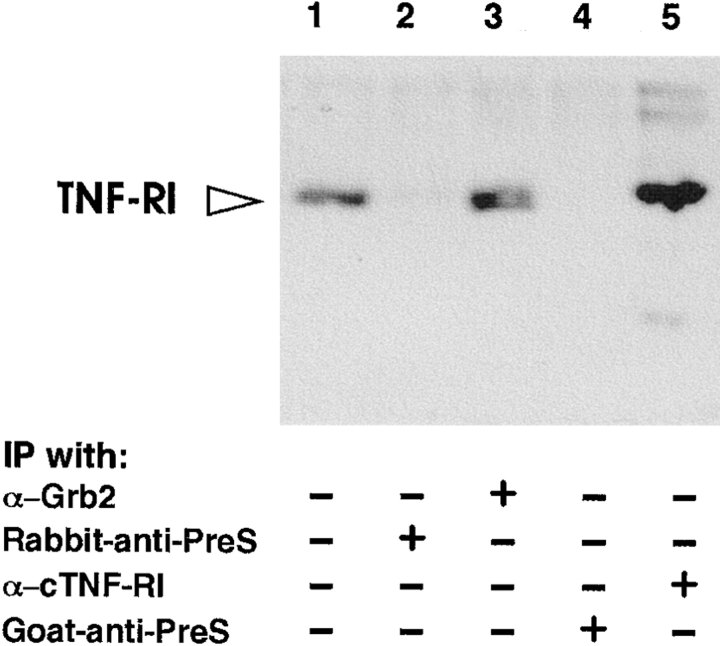
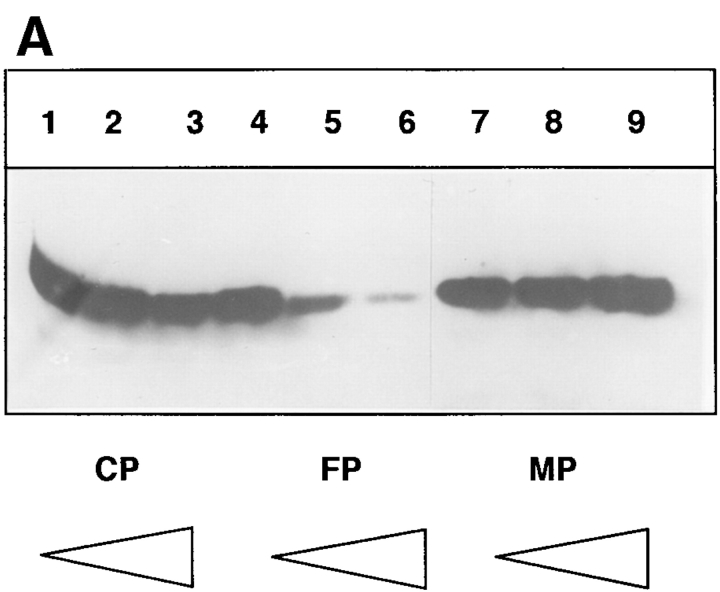
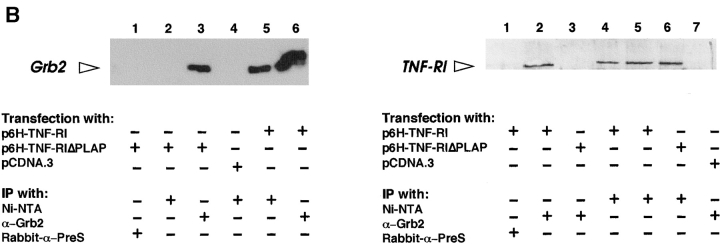
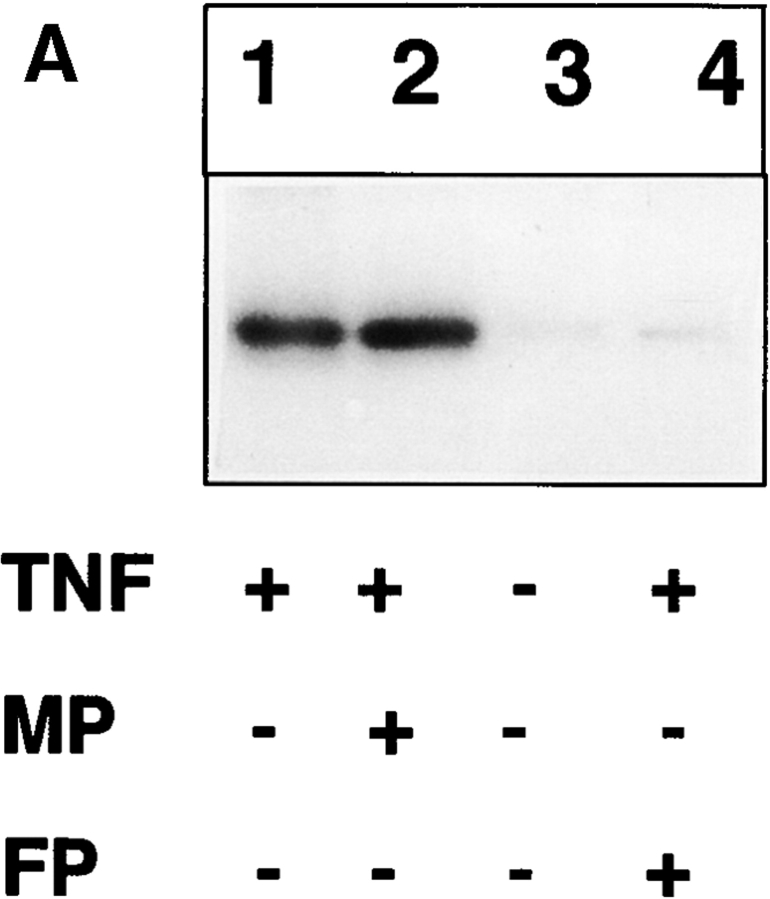
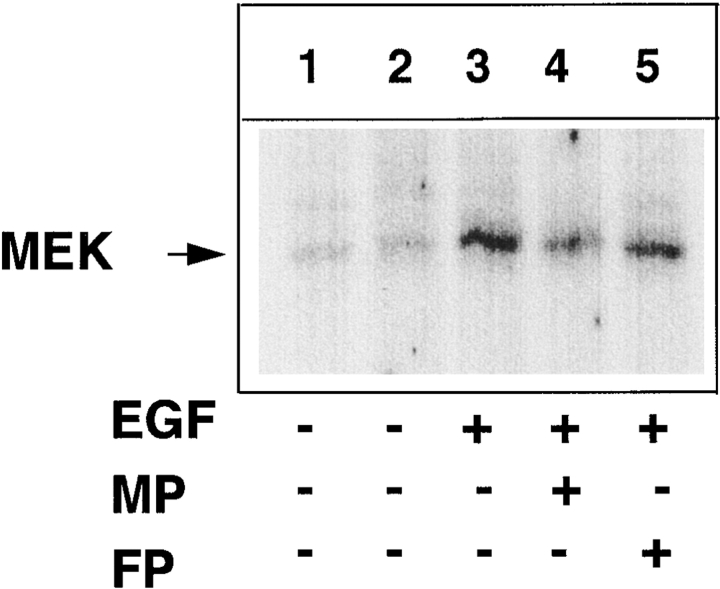
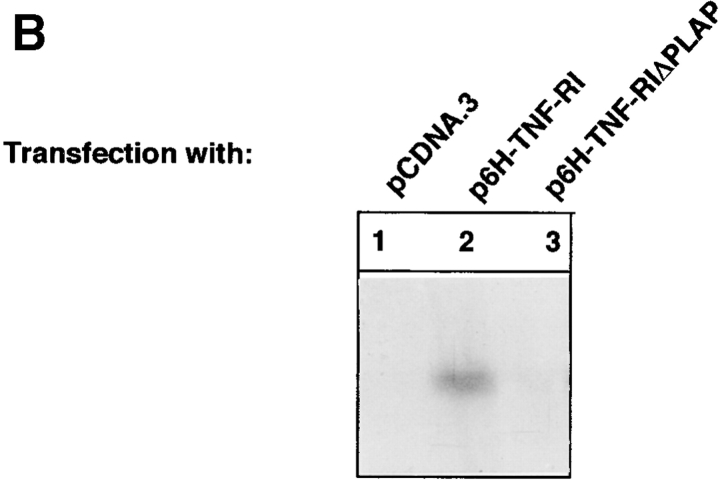
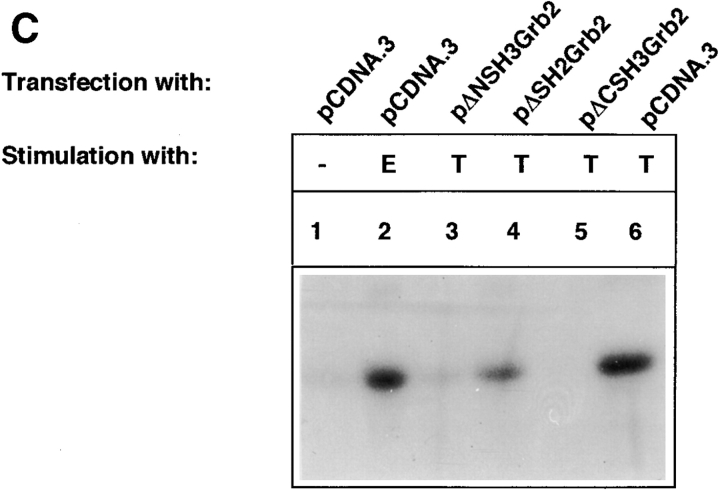
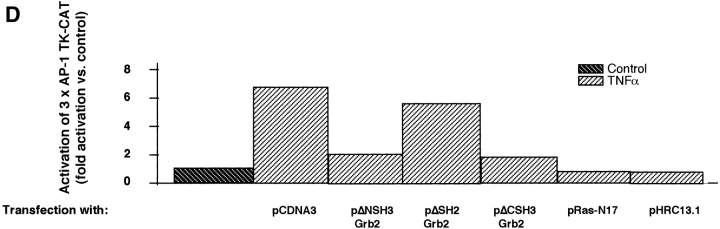
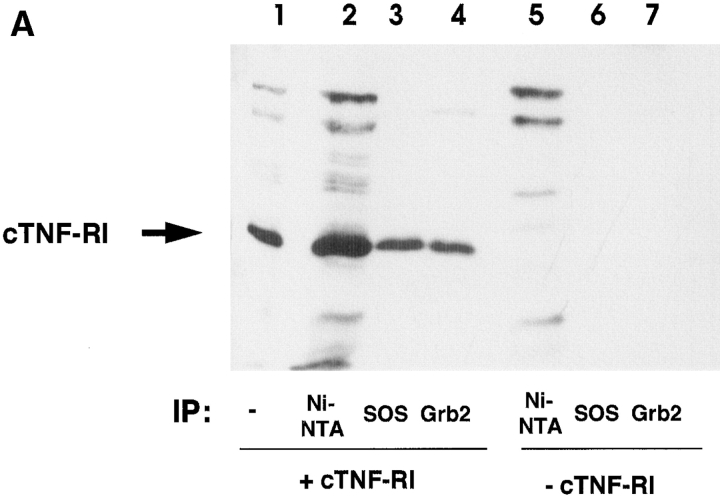
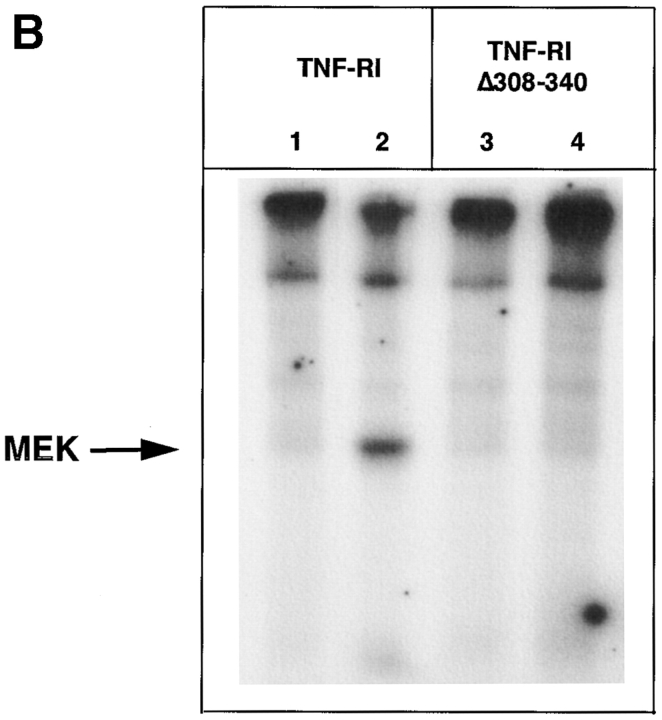
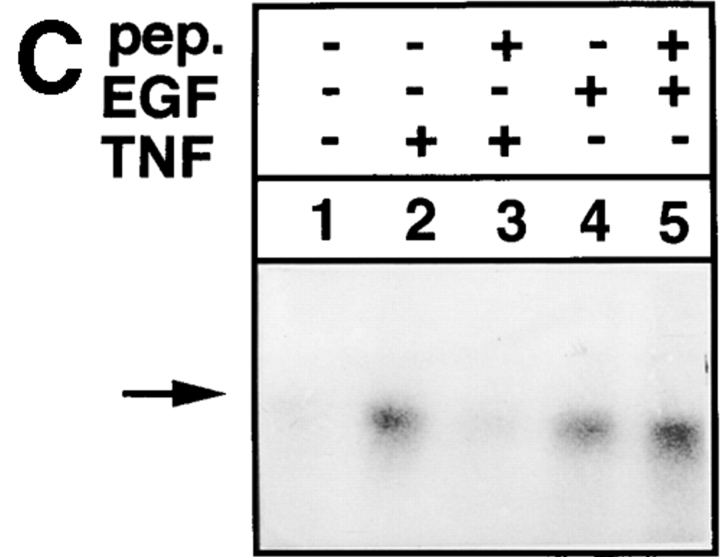
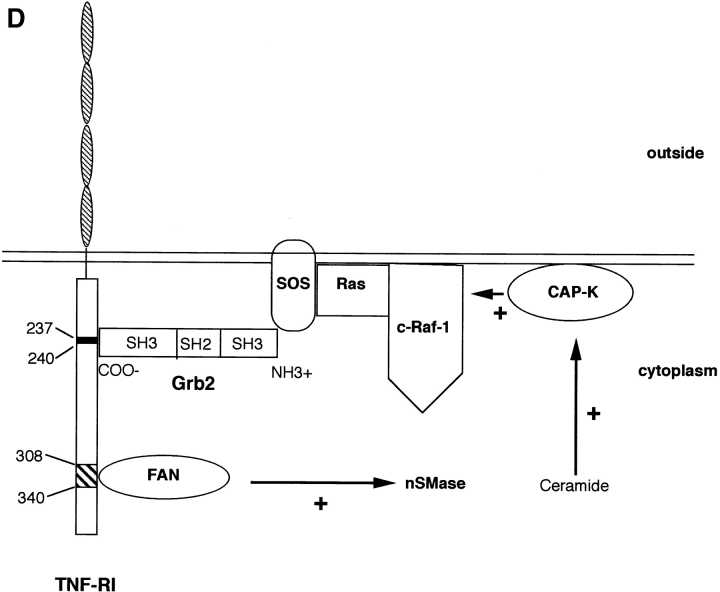
Tumor necrosis factor alpha (TNF-alpha) is a proinflammatory cytokine. Its pleiotropic biological properties are signaled through two distinct cell surface receptors: the TNF receptor type I (TNFR-I) and the TNF receptor type II. Neither of the two receptors possesses tyrosine kinase activity. A large majority of TNF-alpha-dependent activities can be mediated by TNFR-I. Recently, c-Raf-1 kinase was identified as an intracellular target of a signal transduction cascade initiated by binding of TNF-alpha to TNFR-I. However, the mechanism engaged in TNF-alpha-dependent activation of c-Raf-1 kinase is still enigmatic. Here we report that the cytosolic adapter protein Grb2 is a novel binding partner of TNFR-I. Grb2 binds with its COOH-terminal SH3 domain to a PLAP motif within TNFR-I and with its NH2-terminal SH3 domain to SOS (son of sevenless). A PLAP deletion mutant of TNFR-I fails to bind Grb2. The TNFR-I/Grb2 interaction is essential for the TNF-alpha-dependent activation of c-Raf-1 kinase; activation of c-Raf-1 kinase by TNF-alpha can be blocked by coexpression of Grb2 mutants harboring inactivating point mutations in the NH2- or COOH-terminal SH3 domain, cell-permeable peptides that disrupt the Grb2/TNFR-I interaction or transdominant negative Ras. Functionality of the TNFR-I/Grb2/SOS/Ras interaction is a prerequisite but not sufficient for TNF-alpha-dependent activation of c-Raf-1 kinase. Inhibition of the TNFR-I/FAN (factor associated with neutral sphingomyelinase) interaction, which is essential for TNF-alpha-dependent activation of the neutral sphingomyelinase, either by cell-permeable peptides or by deletion of the FAN binding domain, prevents activation of c-Raf-1 kinase. In conclusion, binding of the Grb2 adapter protein via its COOH-terminal SH3 domain to the nontyrosine kinase receptor TNFR-I results in activation of a signaling cascade known so far to be initiated, in the case of the tyrosine kinase receptors, by binding of the SH2 domain of Grb2 to phosphotyrosine.
Figures
Similar articles
-
Tight association of GRB2 with receptor protein-tyrosine phosphatase alpha is mediated by the SH2 and C-terminal SH3 domains.EMBO J. 1996 Jun 17;15(12):3016-27. EMBO J. 1996. PMID: 8670803 Free PMC article.
-
Grb2 SH3 binding to peptides from Sos: evaluation of a general model for SH3-ligand interactions.Chem Biol. 1995 Jan;2(1):53-60. doi: 10.1016/1074-5521(95)90080-2. Chem Biol. 1995. PMID: 9383403
-
SH3 domains of the adapter molecule Grb2 complex with two proteins in T cells: the guanine nucleotide exchange protein Sos and a 75-kDa protein that is a substrate for T cell antigen receptor-activated tyrosine kinases.J Biol Chem. 1994 May 13;269(19):14081-7. J Biol Chem. 1994. PMID: 8188688
-
Distinct adapter proteins mediate acid versus neutral sphingomyelinase activation through the p55 receptor for tumor necrosis factor.J Leukoc Biol. 1998 Jun;63(6):678-82. doi: 10.1002/jlb.63.6.678. J Leukoc Biol. 1998. PMID: 9620659 Review.
-
Inhibitors of Ras signal transduction as antitumor agents.Biochem Pharmacol. 2000 Oct 15;60(8):1165-9. doi: 10.1016/s0006-2952(00)00428-7. Biochem Pharmacol. 2000. PMID: 11007954 Review.
Cited by
-
The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice.EMBO J. 2002 Feb 15;21(4):525-35. doi: 10.1093/emboj/21.4.525. EMBO J. 2002. PMID: 11847101 Free PMC article.
-
TRUSS, a novel tumor necrosis factor receptor 1 scaffolding protein that mediates activation of the transcription factor NF-kappaB.Mol Cell Biol. 2003 Nov;23(22):8334-44. doi: 10.1128/MCB.23.22.8334-8344.2003. Mol Cell Biol. 2003. PMID: 14585990 Free PMC article.
-
The EGF receptor and HER2 participate in TNF-α-dependent MAPK activation and IL-8 secretion in intestinal epithelial cells.Mediators Inflamm. 2012;2012:207398. doi: 10.1155/2012/207398. Epub 2012 Sep 5. Mediators Inflamm. 2012. PMID: 22988345 Free PMC article.
-
Prioritizing candidate disease genes by network-based boosting of genome-wide association data.Genome Res. 2011 Jul;21(7):1109-21. doi: 10.1101/gr.118992.110. Epub 2011 May 2. Genome Res. 2011. PMID: 21536720 Free PMC article.
-
TNF-induced signaling in apoptosis.J Clin Immunol. 1999 Nov;19(6):350-64. doi: 10.1023/a:1020546615229. J Clin Immunol. 1999. PMID: 10634209 Review.
References
-
- Fiers W. Tumor necrosis factor; characterization at the molecular, cellular and in vivo level. FEBS (Fed Eur Biochem Soc) Lett. 1991;285:199–212. - PubMed
-
- Schoenfeldt HJ, Poeschl B, Frey JR, Loetscher J, Hunziker W, Lustig A, Zulauf M. Efficient purification of recombinant human tumor necrosis factor beta from E. coliyields biological active protein with a trimeteric structure that binds to both tumor necrosis factor receptors. J Biol Chem. 1991;266:3863–3869. - PubMed
-
- Bazzoni, R., and T. Beuteler. 1996. The tumor necrosis factor ligand and receptor families. In Cytokines. J.S. Flier, editor. Massachusetts Medical Society, Boston, MA. 334:1717– 1725. - PubMed
-
- Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kroenke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenesinfection. Cell. 1993;73:457–467. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
