

An estrogen receptor-selective coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens
- PMID: 10359819
- PMCID: PMC22022
- DOI: 10.1073/pnas.96.12.6947
An estrogen receptor-selective coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens
Abstract
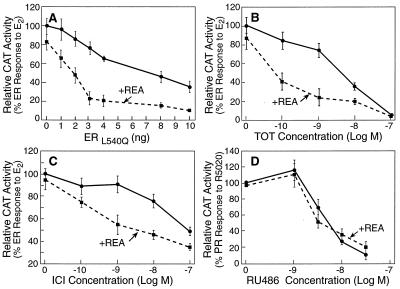
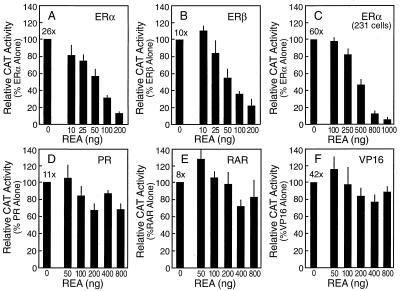
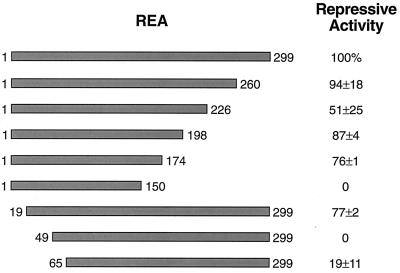
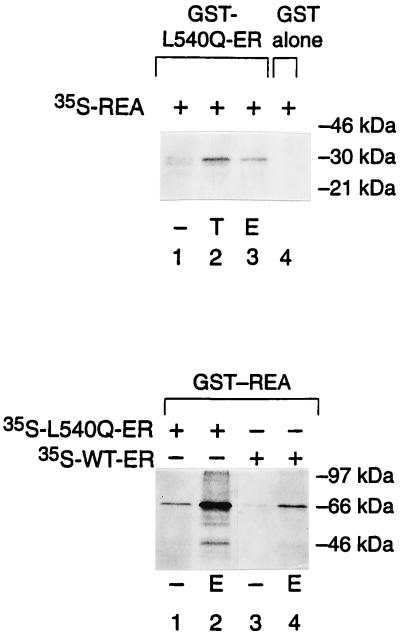
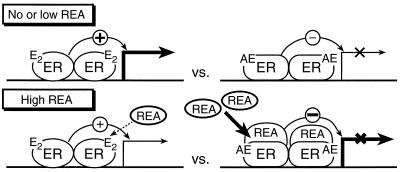
The action of nuclear hormone receptors is tripartite, involving the receptor, its ligands, and its coregulator proteins. The estrogen receptor (ER), a member of this superfamily, is a hormone-activated transcription factor that mediates the stimulatory effects of estrogens and the inhibitory effects of antiestrogens such as tamoxifen in breast cancer and other estrogen target cells. To understand how antiestrogens and dominant negative ERs suppress ER activity, we used a dominant negative ER as bait in two-hybrid screening assays from which we isolated a clone from breast cancer cells that potentiates the inhibitory activities of dominant negative ERs and antiestrogen-liganded ER. At higher concentrations, it also represses the transcriptional activity of the estradiol-liganded ER, while having no effect on other nuclear hormone receptors. This clone, denoted REA for "repressor of estrogen receptor activity," encodes a 37-kDa protein that is an ER-selective coregulator. Its competitive reversal of steroid receptor coactivator 1 enhancement of ER activity and its direct interaction with liganded ER suggest that it may play an important role in determining the sensitivity of estrogen target cells, including breast cancer cells, to antiestrogens and estrogens.
Figures


References
-
- Katzenellenbogen J A, O’Malley B W, Katzenellenbogen B S. Mol Endocrinol. 1996;10:119–131. - PubMed
-
- Shibata H, Spencer T E, Onate S A, Jenster G, Tsai S Y, Tsai M-J, O’Mally B W. Recent Prog Horm Res. 1997;52:141–165. - PubMed
-
- Horwitz K B, Jackson T A, Bain D L, Richer J K, Takimoto G S. Mol Endocrinol. 1996;10:1167–1177. - PubMed
-
- Katzenellenbogen B S, Montano M M, Ekena K, Herman M E, McInerney E M. Breast Cancer Res Treat. 1997;44:23–38. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
