

Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice
- PMID: 10359843
- PMCID: PMC22065
- DOI: 10.1073/pnas.96.12.7088
Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice
Abstract
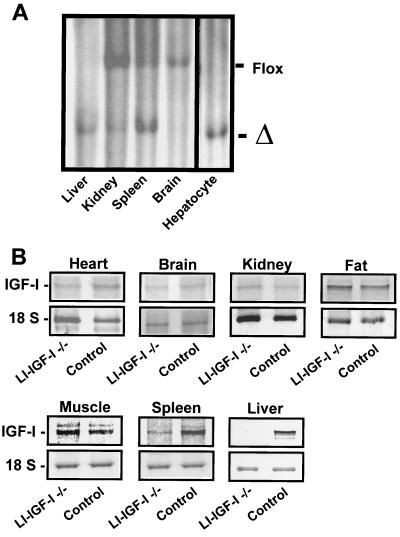
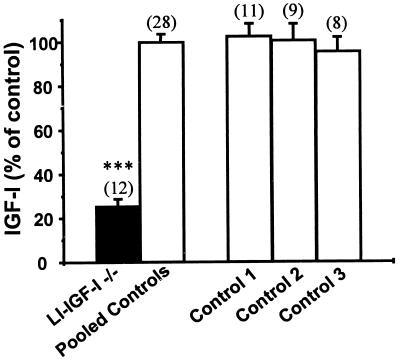
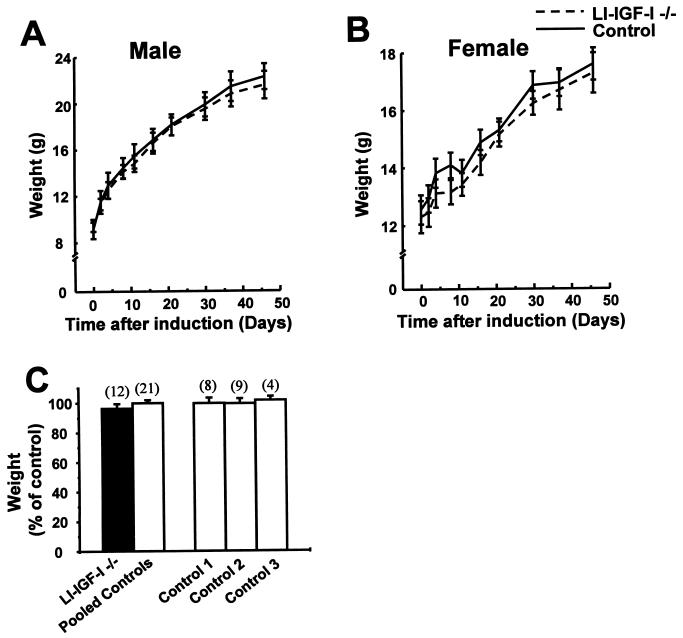
The body growth of animals is regulated by growth hormone and IGF-I. The classical theory of this regulation is that most IGF-I in the blood originates in the liver and that body growth is controlled by the concentration of IGF-I in the blood. We have abolished IGF-I production in the livers of mice by using the Cre/loxP recombination system. These mice demonstrated complete inactivation of the IGF-I gene in the hepatocytes. Although the liver accounts for less than 5% of body mass, the concentration of IGF-I in the serum was reduced by 75%. This finding confirms that the liver is the principal source of IGF-I in the blood. However, the reduction in serum IGF-I concentration had no discernible effect on postnatal body growth. We conclude that postnatal body growth is preserved despite complete absence of IGF-I production by the hepatocytes.
Figures
Similar articles
-
Effects of growth hormone and insulinlike growth factor-I on body growth and adult bone metabolism.Curr Opin Rheumatol. 2000 Jul;12(4):346-8. doi: 10.1097/00002281-200007000-00019. Curr Opin Rheumatol. 2000. PMID: 10910189 Review.
-
Age-dependent onset of liver-specific IGF-I gene deficiency and its persistence in old age: implications for postnatal growth and insulin resistance in LID mice.Am J Physiol Endocrinol Metab. 2005 Aug;289(2):E288-95. doi: 10.1152/ajpendo.00494.2004. Epub 2005 Mar 15. Am J Physiol Endocrinol Metab. 2005. PMID: 15769793
-
Insulin-like growth factor I is essential for postnatal growth in response to growth hormone.Endocrinology. 1999 Nov;140(11):5178-84. doi: 10.1210/endo.140.11.7151. Endocrinology. 1999. PMID: 10537147
-
The relative importance of endocrine versus autocrine/paracrine insulin-like growth factor-I in the regulation of body growth.Pediatr Nephrol. 2000 Jul;14(7):541-3. doi: 10.1007/s004670000348. Pediatr Nephrol. 2000. PMID: 10912515 Review.
-
Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system.Proc Soc Exp Biol Med. 2000 Apr;223(4):344-51. doi: 10.1046/j.1525-1373.2000.22349.x. Proc Soc Exp Biol Med. 2000. PMID: 10721003 Review.
Cited by
-
A botulinum toxin-derived targeted secretion inhibitor downregulates the GH/IGF1 axis.J Clin Invest. 2012 Sep;122(9):3295-306. doi: 10.1172/JCI63232. Epub 2012 Aug 1. J Clin Invest. 2012. PMID: 22850878 Free PMC article.
-
Excitation-transcription coupling in skeletal muscle: the molecular pathways of exercise.Biol Rev Camb Philos Soc. 2011 Aug;86(3):564-600. doi: 10.1111/j.1469-185X.2010.00161.x. Epub 2010 Oct 6. Biol Rev Camb Philos Soc. 2011. PMID: 21040371 Free PMC article. Review.
-
Deletion of IGF-I receptor (IGF-IR) in primary osteoblasts reduces GH-induced STAT5 signaling.Mol Endocrinol. 2010 Mar;24(3):644-56. doi: 10.1210/me.2009-0357. Epub 2010 Feb 4. Mol Endocrinol. 2010. PMID: 20133448 Free PMC article.
-
Tissue-specific disruption of the growth hormone receptor (GHR) in mice: An update.Growth Horm IGF Res. 2020 Apr;51:1-5. doi: 10.1016/j.ghir.2019.12.004. Epub 2019 Dec 24. Growth Horm IGF Res. 2020. PMID: 31923746 Free PMC article. Review.
-
The decrease in mature myostatin protein in male skeletal muscle is developmentally regulated by growth hormone.J Physiol. 2009 Feb 1;587(3):669-77. doi: 10.1113/jphysiol.2008.161521. Epub 2008 Dec 1. J Physiol. 2009. PMID: 19047209 Free PMC article.
References
-
- Ohlsson C, Bengtsson B A, Isaksson O G, Andreassen T T, Slootweg M C. Endocr Rev. 1998;19:55–79. - PubMed
-
- Isaksson O G, Lindahl A, Nilsson A, Isgaard J. Endocr Rev. 1987;8:426–438. - PubMed
-
- Daughaday W H, Rotwein P. Endocr Rev. 1989;10:68–91. - PubMed
-
- Holder A T, Spencer E M, Preece M A. J Endocrinol. 1981;89:275–282. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
