

The genomic tree as revealed from whole proteome comparisons
- PMID: 10400922
- PMCID: PMC310764
The genomic tree as revealed from whole proteome comparisons
Abstract
The availability of a number of complete cellular genome sequences allows the development of organisms' classification, taking into account their genome content, the loss or acquisition of genes, and overall gene similarities as signatures of common ancestry. On the basis of correspondence analysis and hierarchical classification methods, a methodological framework is introduced here for the classification of the available 20 completely sequenced genomes and partial information for Schizosaccharomyces pombe, Homo sapiens, and Mus musculus. The outcome of such an analysis leads to a classification of genomes that we call a genomic tree. Although these trees are phenograms, they carry with them strong phylogenetic signatures and are remarkably similar to 16S-like rRNA-based phylogenies. Our results suggest that duplication and deletion events that took place through evolutionary time were globally similar in related organisms. The genomic trees presented here place the Archaea in the proximity of the Bacteria when the whole gene content of each organism is considered, and when ancestral gene duplications are eliminated. Genomic trees represent an additional approach for the understanding of evolution at the genomic level and may contribute to the proper assessment of the evolutionary relationships between extant species.
Figures
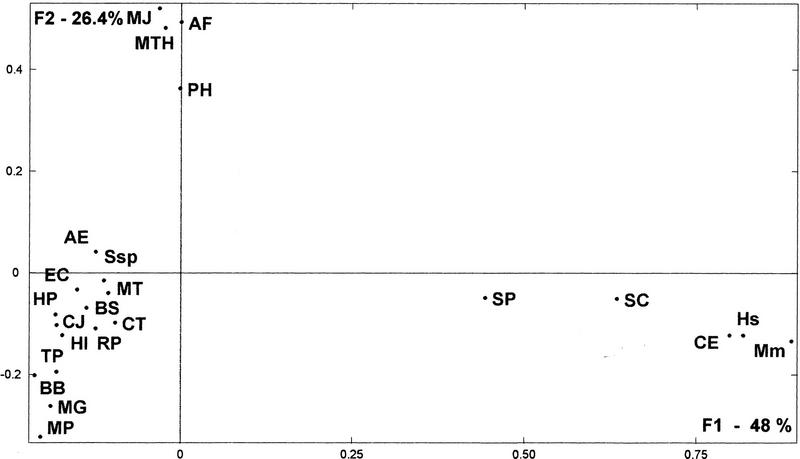
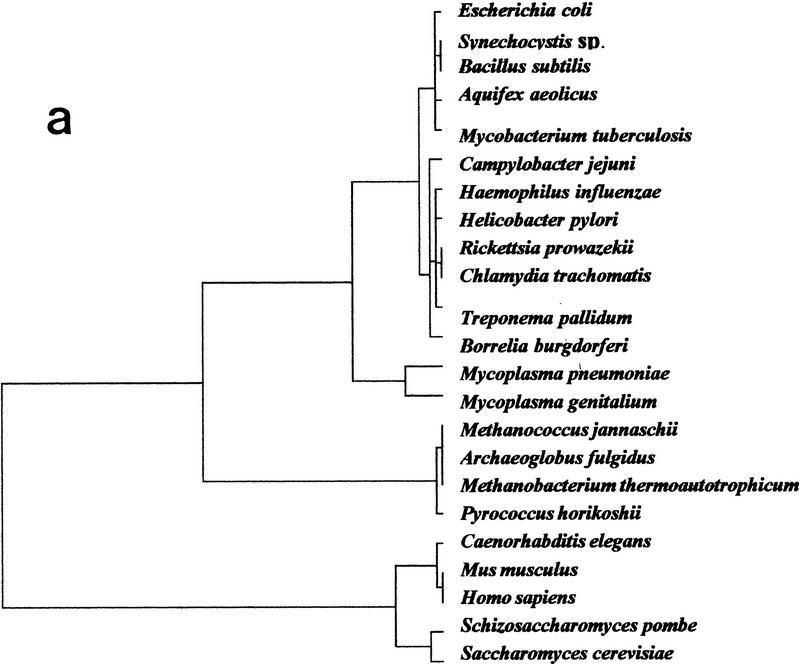
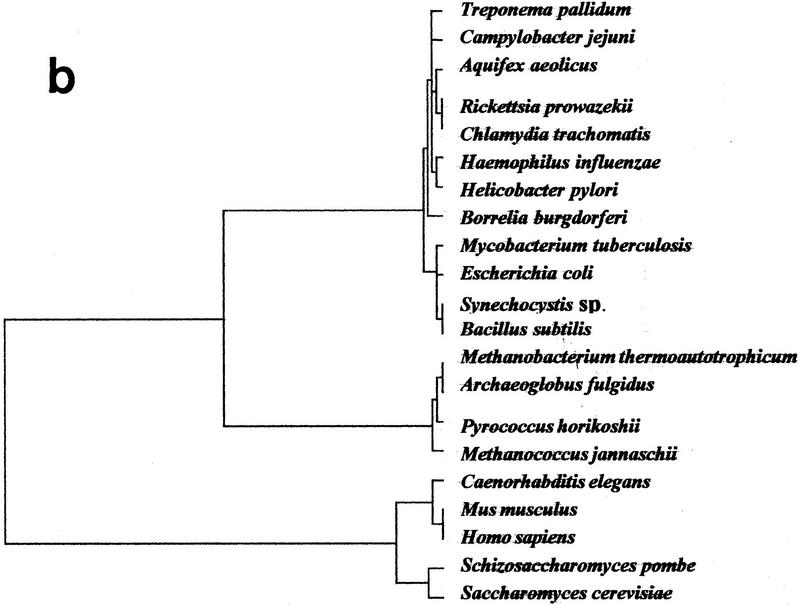
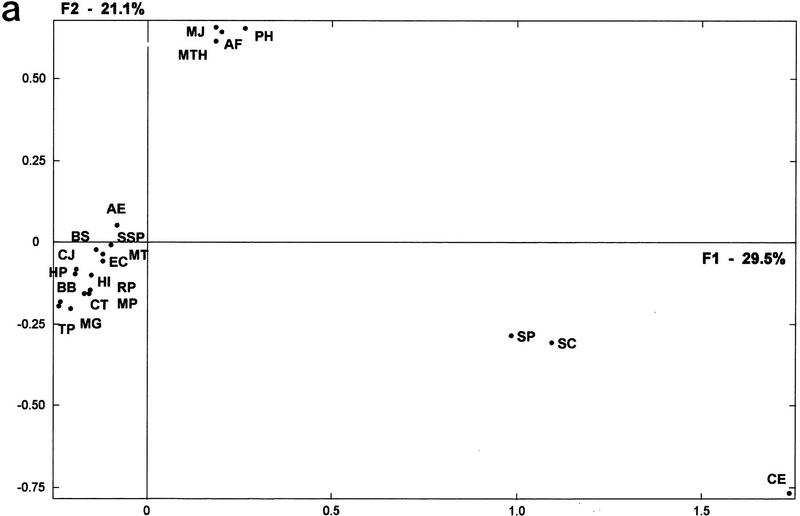
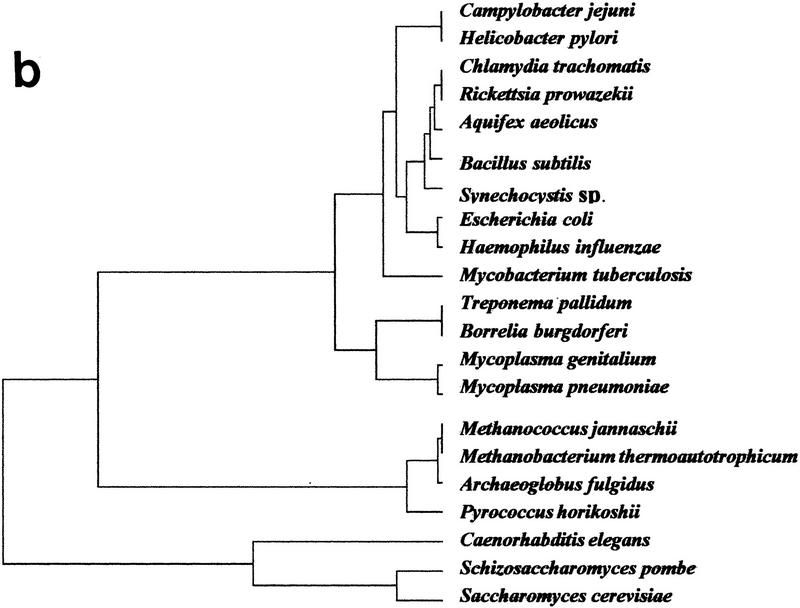
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. - PubMed
-
- Benzecri J-P. L’analyse des données. Vol 2: L’analyse des correspondances. Paris, France: Dunod; 1973.
-
- Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. - PubMed
-
- Boguski MS, Schuler GD. Establishing a human transcript map. Nat Genet. 1995;10:369–371. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous