

Differentiation-induced internal translation of c-sis mRNA: analysis of the cis elements and their differentiation-linked binding to the hnRNP C protein
- PMID: 10409733
- PMCID: PMC84385
- DOI: 10.1128/MCB.19.8.5429
Differentiation-induced internal translation of c-sis mRNA: analysis of the cis elements and their differentiation-linked binding to the hnRNP C protein
Abstract
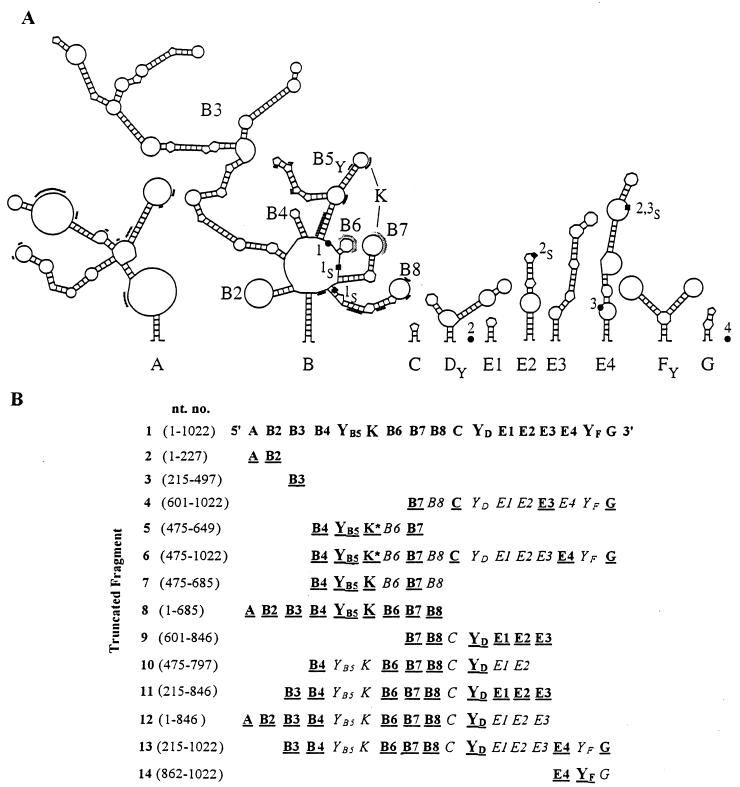
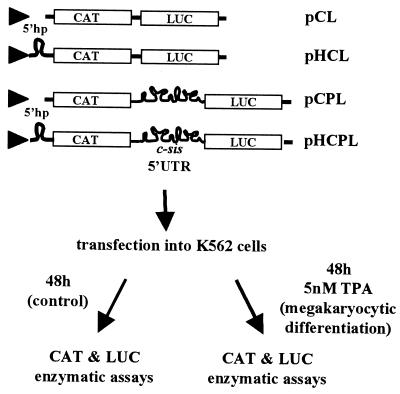
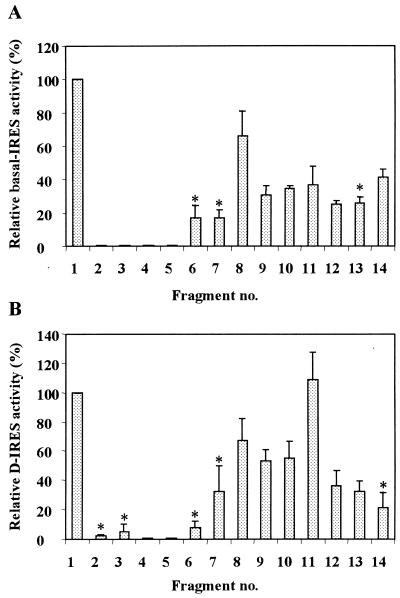
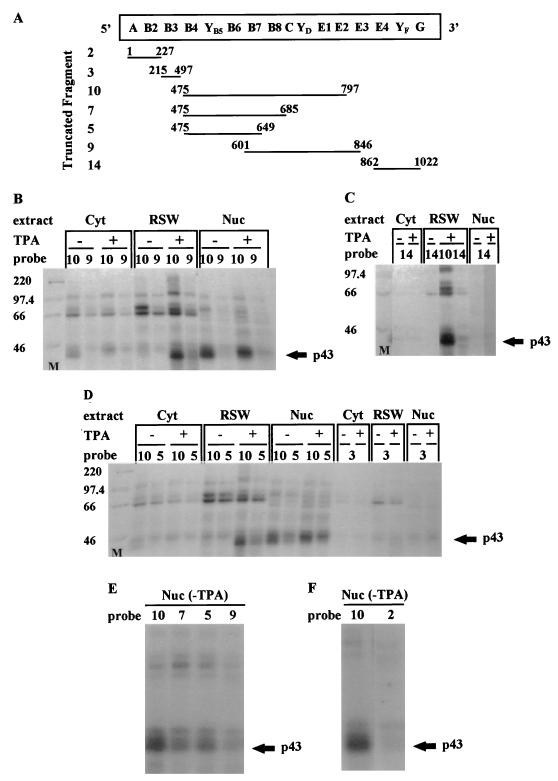
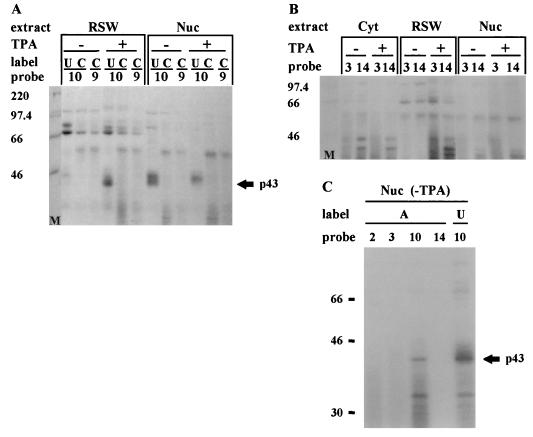
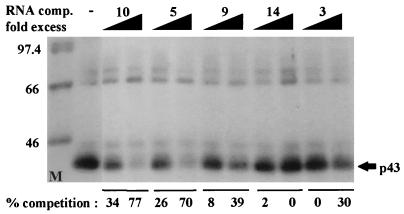
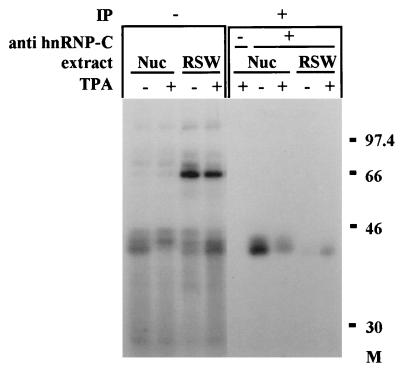
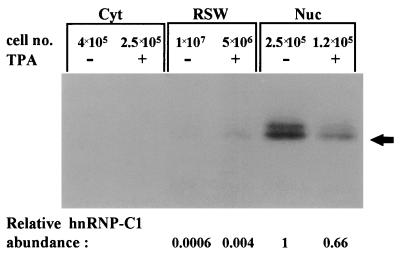
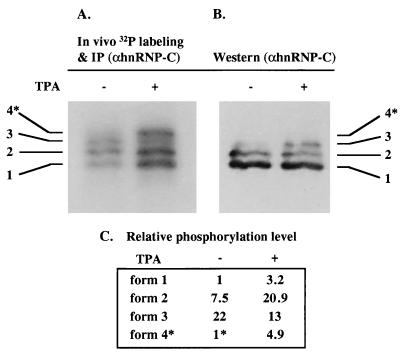
In previous reports we showed that the long 5' untranslated region (5' UTR) of c-sis, the gene encoding the B chain of platelet-derived growth factor, has translational modulating activity due to its differentiation-activated internal ribosomal entry site (D-IRES). Here we show that the 5' UTR contains three regions with a computer-predicted Y-shaped structure upstream of an AUG codon, each of which can confer some degree of internal translation by itself. In nondifferentiated cells, the entire 5' UTR is required for maximal basal IRES activity. The elements required for the differentiation-sensing ability (i.e., D-IRES) were mapped to a 630-nucleotide fragment within the central portion of the 5' UTR. Even though the region responsible for IRES activation is smaller, the full-length 5' UTR is capable of mediating the maximal translation efficiency in differentiated cells, since only the entire 5' UTR is able to confer the maximal basal IRES activity. Interestingly, a 43-kDa protein, identified as hnRNP C, binds in a differentiation-induced manner to the differentiation-sensing region. Using UV cross-linking experiments, we show that while hnRNP C is mainly a nuclear protein, its binding activity to the D-IRES is mostly nuclear in nondifferentiated cells, whereas in differentiated cells such binding activity is associated with the ribosomal fraction. Since the c-sis 5' UTR is a translational modulator in response to cellular changes, it seems that the large number of cross-talking structural entities and the interactions with regulated trans-acting factors are important for the strength of modulation in response to cellular changes. These characteristics may constitute the major difference between strong IRESs, such as those seen in some viruses, and IRESs that serve as translational modulators in response to developmental signals, such as that of c-sis.
Figures
Similar articles
-
Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms.Mol Cell Biol. 2000 Apr;20(7):2297-307. doi: 10.1128/MCB.20.7.2297-2307.2000. Mol Cell Biol. 2000. PMID: 10713153 Free PMC article.
-
Tight control of platelet-derived growth factor B/c-sis expression by interplay between the 5'-untranslated region sequence and the major upstream promoter.J Biol Chem. 2003 Nov 21;278(47):46983-93. doi: 10.1074/jbc.M304976200. Epub 2003 Sep 5. J Biol Chem. 2003. PMID: 12960151
-
PDGF2/c-sis mRNA leader contains a differentiation-linked internal ribosomal entry site (D-IRES).J Biol Chem. 1997 Apr 4;272(14):9356-62. doi: 10.1074/jbc.272.14.9356. J Biol Chem. 1997. PMID: 9083072
-
The role of the 5' untranslated region of an mRNA in translation regulation during development.Int J Biochem Cell Biol. 1999 Jan;31(1):87-106. doi: 10.1016/s1357-2725(98)00134-4. Int J Biochem Cell Biol. 1999. PMID: 10216946 Review.
-
An atypical IRES within the 5' UTR of a dicistrovirus genome.Virus Res. 2009 Feb;139(2):157-65. doi: 10.1016/j.virusres.2008.07.017. Epub 2008 Sep 11. Virus Res. 2009. PMID: 18755228 Review.
Cited by
-
Dicistronic MLV-retroviral vectors transduce neural precursors in vivo and co-express two genes in their differentiated neuronal progeny.Retrovirology. 2005 Sep 29;2:60. doi: 10.1186/1742-4690-2-60. Retrovirology. 2005. PMID: 16194277 Free PMC article.
-
Protein kinase CK1alphaLS promotes vascular cell proliferation and intimal hyperplasia.Am J Pathol. 2010 Sep;177(3):1562-72. doi: 10.2353/ajpath.2010.100327. Epub 2010 Aug 9. Am J Pathol. 2010. PMID: 20696773 Free PMC article.
-
Bridging IRES elements in mRNAs to the eukaryotic translation apparatus.Biochim Biophys Acta. 2009 Sep-Oct;1789(9-10):518-28. doi: 10.1016/j.bbagrm.2009.07.004. Epub 2009 Jul 23. Biochim Biophys Acta. 2009. PMID: 19631772 Free PMC article. Review.
-
Suppression of cap-dependent translation in mitosis.Genes Dev. 2001 Aug 15;15(16):2083-93. doi: 10.1101/gad.889201. Genes Dev. 2001. PMID: 11511540 Free PMC article.
-
Identification of two short internal ribosome entry sites selected from libraries of random oligonucleotides.Proc Natl Acad Sci U S A. 2001 Feb 13;98(4):1471-6. doi: 10.1073/pnas.98.4.1471. Proc Natl Acad Sci U S A. 2001. PMID: 11171975 Free PMC article.
References
-
- Akiri G, Nahari D, Finkelstein Y, Le S-Y, Elroy-Stein O, Levi B-Z. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene. 1998;17:227–237. - PubMed
-
- Bernstein J, Shefler I, Elroy-Stein O. The translational repression mediated by the platelet-derived growth factor 2/c-sis mRNA leader is relieved during megakaryocytic differentiation. J Biol Chem. 1995;270:10559–10565. - PubMed
-
- Bernstein J, Sella O, Le S-Y, Elroy-Stein O. PDGF2/c-sis mRNA leader contains a differentiation-linked internal ribosomal entry site (D-IRES) J Biol Chem. 1997;272:9356–9362. - PubMed
-
- Biamonti G, Bassi M T, Cartegni L, Mechta F, Uuvoli M, Cobianchi F, Riva S. Human hnRNP protein A1 gene expression—structural and functional characterization of the promoter. J Mol Biol. 1993;230:77–89. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources