

NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1
- PMID: 10409765
- PMCID: PMC84428
- DOI: 10.1128/MCB.19.8.5785
NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1
Abstract
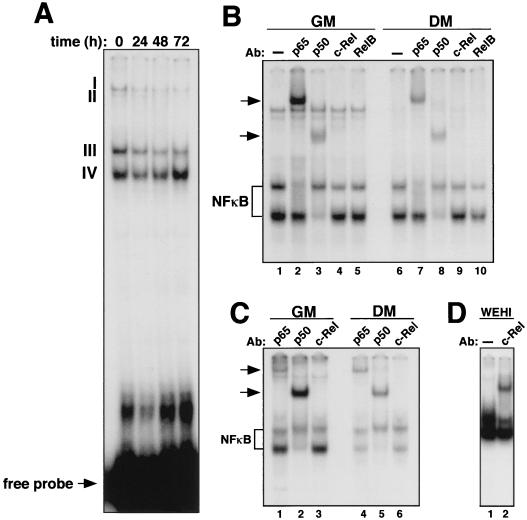
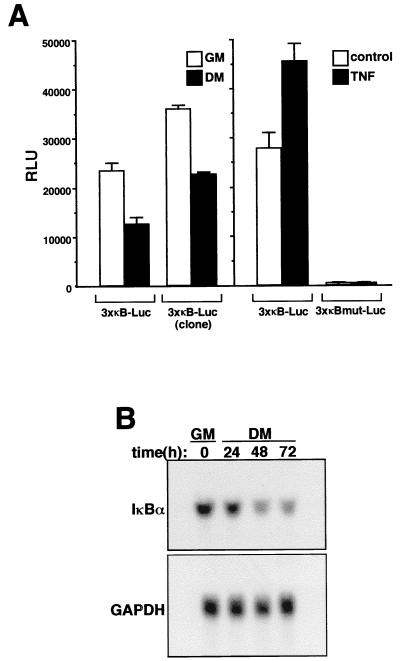
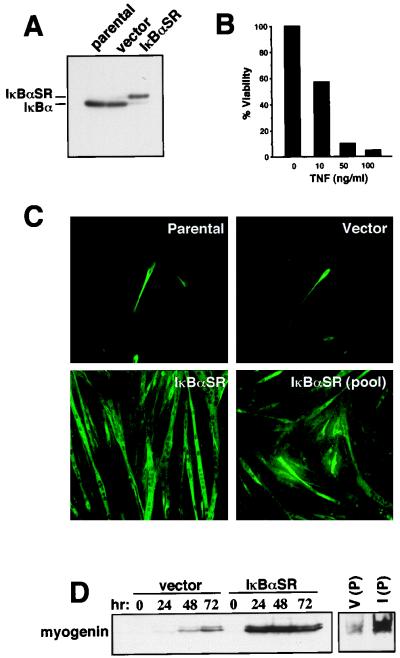
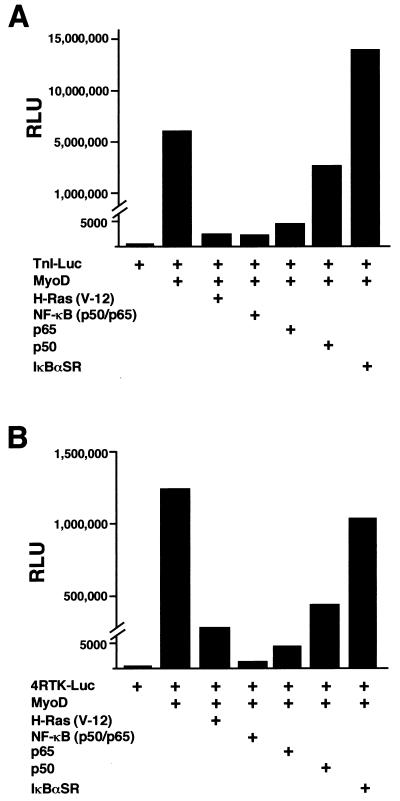
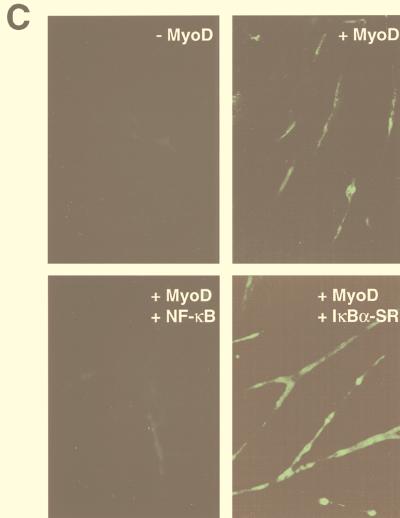
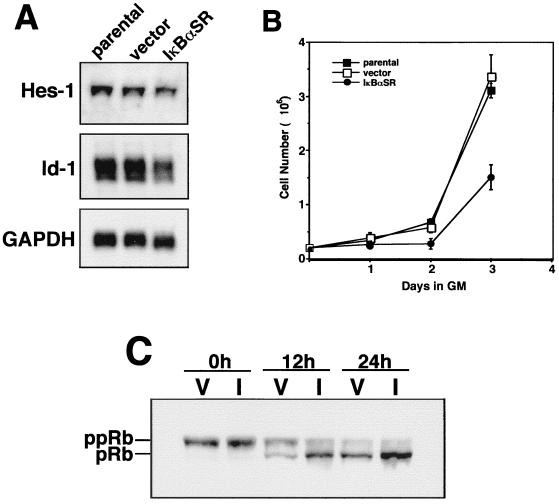
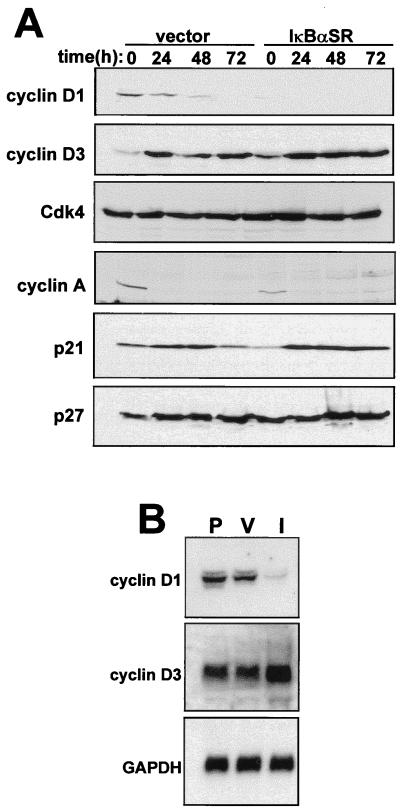
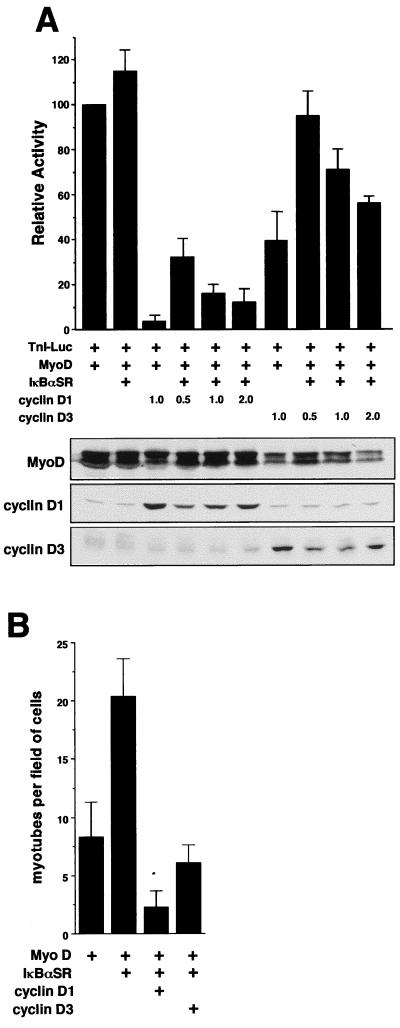
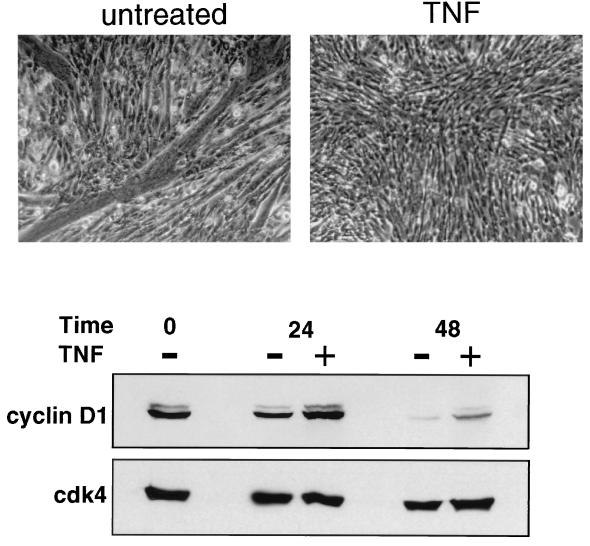
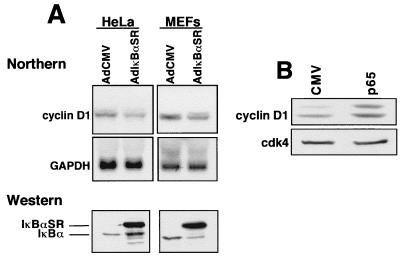
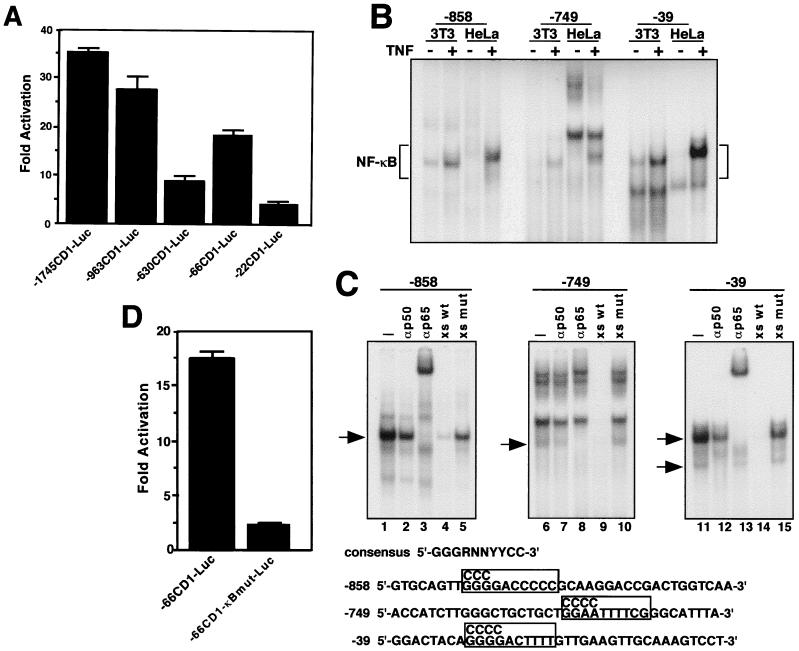
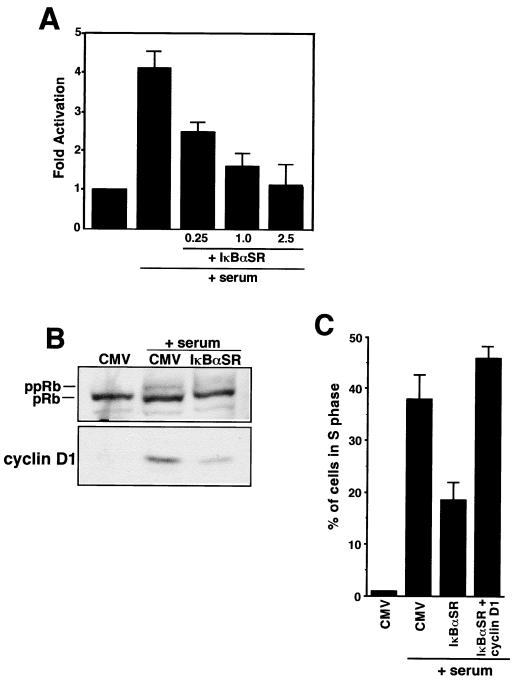
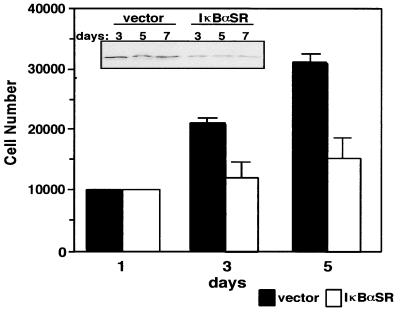
Accumulating evidence implicates the transcription factor NF-kappaB as a positive mediator of cell growth, but the molecular mechanism(s) involved in this process remains largely unknown. Here we use both a skeletal muscle differentiation model and normal diploid fibroblasts to gain insight into how NF-kappaB regulates cell growth and differentiation. Results obtained with the C2C12 myoblast cell line demonstrate that NF-kappaB functions as an inhibitor of myogenic differentiation. Myoblasts generated to lack NF-kappaB activity displayed defects in cellular proliferation and cell cycle exit upon differentiation. An analysis of cell cycle markers revealed that NF-kappaB activates cyclin D1 expression, and the results showed that this regulatory pathway is one mechanism by which NF-kappaB inhibits myogenesis. NF-kappaB regulation of cyclin D1 occurs at the transcriptional level and is mediated by direct binding of NF-kappaB to multiple sites in the cyclin D1 promoter. Using diploid fibroblasts, we demonstrate that NF-kappaB is required to induce cyclin D1 expression and pRb hyperphosphorylation and promote G(1)-to-S progression. Consistent with results obtained with the C2C12 differentiation model, we show that NF-kappaB also promotes cell growth in embryonic fibroblasts, correlating with its regulation of cyclin D1. These data therefore identify cyclin D1 as an important transcriptional target of NF-kappaB and reveal a mechanism to explain how NF-kappaB is involved in the early phases of the cell cycle to regulate cell growth and differentiation.
Figures
References
-
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell R G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. - PubMed
-
- Arber N, Sutter T, Miyake M, Kahn S M, Venkatraj V S, Sobrino A, Warburton D, Holt P R, Weinstein I B. Increased expression of cyclin D1 and the Rb tumor suppressor gene in c-K-ras transformed rat enterocytes. Oncogene. 1996;12:1903–1908. - PubMed
-
- Baeuerle P A, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. - PubMed
-
- Baeuerle P A, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. - PubMed
-
- Baldwin A S., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials