

Generation of influenza A viruses entirely from cloned cDNAs
- PMID: 10430945
- PMCID: PMC17785
- DOI: 10.1073/pnas.96.16.9345
Generation of influenza A viruses entirely from cloned cDNAs
Abstract
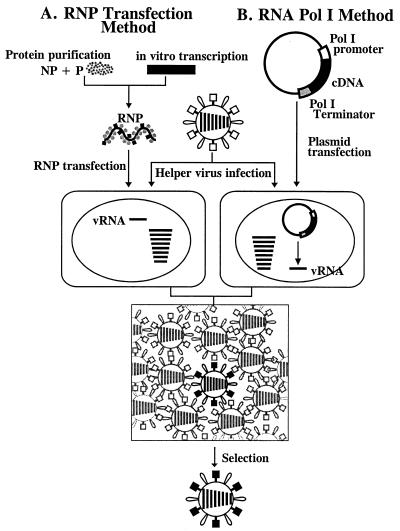
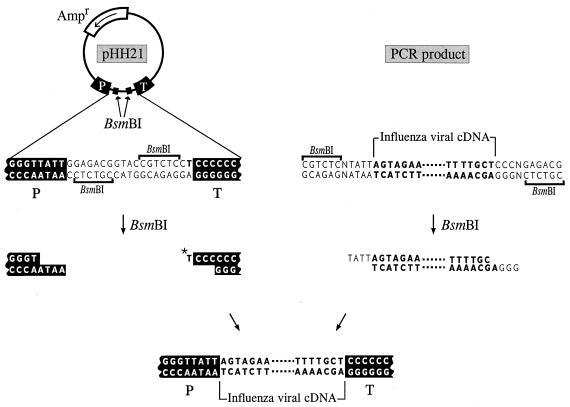
We describe a new reverse-genetics system that allows one to efficiently generate influenza A viruses entirely from cloned cDNAs. Human embryonic kidney cells (293T) were transfected with eight plasmids, each encoding a viral RNA of the A/WSN/33 (H1N1) or A/PR/8/34 (H1N1) virus, flanked by the human RNA polymerase I promoter and the mouse RNA polymerase I terminator-together with plasmids encoding viral nucleoprotein and the PB2, PB1, and PA viral polymerases. This strategy yielded >1 x 10(3) plaque-forming units (pfu) of virus per ml of supernatant at 48 hr posttransfection. The addition of plasmids expressing all of the remaining viral structural proteins led to a substantial increase in virus production, 3 x 10(4)-5 x 10(7) pfu/ml. We also used reverse genetics to generate a reassortant virus containing the PB1 gene of the A/PR/8/34 virus, with all other genes representing A/WSN/33. Additional viruses produced by this method had mutations in the PA gene or possessed a foreign epitope in the head of the neuraminidase protein. This efficient system, which does not require helper virus infection, should be useful in viral mutagenesis studies and in the production of vaccines and gene therapy vectors.
Figures
Comment in
-
Reverse genetics of negative-strand RNA viruses: closing the circle.Proc Natl Acad Sci U S A. 1999 Aug 3;96(16):8804-6. doi: 10.1073/pnas.96.16.8804. Proc Natl Acad Sci U S A. 1999. PMID: 10430844 Free PMC article. Review. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
