

Pulmonary outcome in cystic fibrosis is influenced primarily by mucoid Pseudomonas aeruginosa infection and immune status and only modestly by genotype
- PMID: 10456926
- PMCID: PMC96804
- DOI: 10.1128/IAI.67.9.4744-4750.1999
Pulmonary outcome in cystic fibrosis is influenced primarily by mucoid Pseudomonas aeruginosa infection and immune status and only modestly by genotype
Abstract
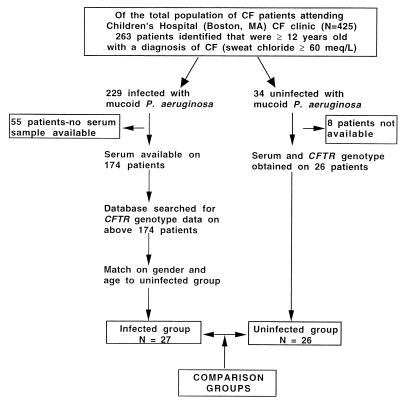
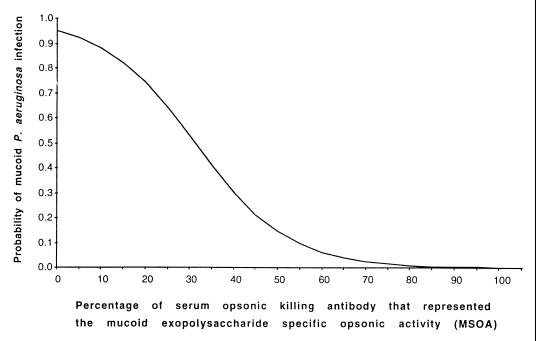
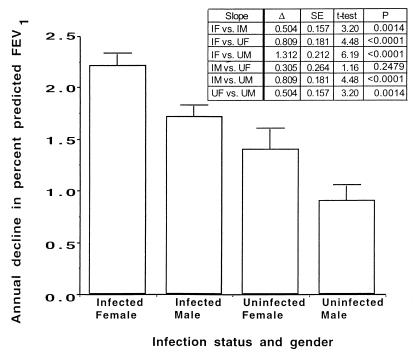
Whether allelic variants of the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) independently contribute to pulmonary outcome in CF patients has not been resolved. We used both cross-sectional and mixed-model longitudinal analyses of data from CF patients that were at least 12 years old to determine the influence on pulmonary function (percent predicted forced expiratory volume [FEV(1)]) of the CFTR gene genotype, gender, mucoid Pseudomonas aeruginosa (MPA) infection status, presence of total opsonic antibody to MPA, and, separately, the opsonic antibody activity specific to the mucoid exopolysaccharide (MEP) surface antigen. Two different factors were independently associated with the lack of MPA infection: a high level of MEP-specific opsonic activity (MSOA), implicating an immunologically based mechanism of resistance to infection, and a lack of any type of opsonic antibody to MPA, indicative of no significant exposure or infection. This latter phenotype was found in a subset of CF patients who carried at least one uncommon CFTR gene allele suggestive of a genetic basis for resistance to infection in this group of older CF patients. For CF patients in whom both CFTR gene alleles were identified by screening for the 12 most common variants (75% of alleles), cross-sectional analysis showed that MPA infection was best correlated with lower percent predicted FEV(1), while genotype (two versus one DeltaF508 CFTR gene allele) and a low level of MSOA were associated with increased risk of infection. A mixed-model analysis of longitudinal spirometric measurements that considered multiple risk factors to derive regression equations was used to determine which clinical parameters had the greatest effect on the annual rate of decline in percent predicted FEV(1). This analysis showed that the CFTR gene genotype only modestly modified the constant (y intercept) of the derived equations, while gender and MPA infection status had the largest effects on annual rates of decline in percent predicted FEV(1). These results indicate that the CFTR genotype is usually not a primary determinant of pulmonary function in most CF patients, but gender and MPA infection status are. Infection status is potentially influenced by both immunologic (a high level of MSOA) and genetic factors, such as carriage of a CFTR gene allele that leads to a diagnosis of CF but still confers resistance to infection that is comparable to that of the wild-type CFTR gene.
Figures
References
-
- Borgo G, Gasparini P, Bonizzato A, Cabrini G, Mastella G, Pignatti P F. Cystic fibrosis: the ΔF508 mutation does not lead to an exceptionally severe phenotype. A cohort study. Eur J Pediatr. 1993;152:1006–1011. - PubMed
-
- Campbell P W, III, Parker R A, Roberts B T, Krishnamani M R, Phillips J A., III Association of poor clinical status and heavy exposure to tobacco smoke in patients with cystic fibrosis who are homozygous for the ΔF508 deletion. J Pediatr. 1992;120:261–264. - PubMed
-
- Campbell P W, III, Phillips III J A, Krishnamani M R, Maness K J, Hazinski T A. Cystic fibrosis: relationship between clinical status and ΔF508 deletion. J Pediatr. 1991;118:239–241. - PubMed
-
- Chillon M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, Romey M C, Ruizromero J, Verlingue C, Claustres M, Nunes V, Ferec C, Estivill X. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–1480. - PubMed
-
- Corey M, Edwards L, Levison H, Knowles M. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr. 1997;131:809–814. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
