

Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones
- PMID: 10457062
- PMCID: PMC2269520
- DOI: 10.1111/j.1469-7793.1999.0451m.x
Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones
Abstract
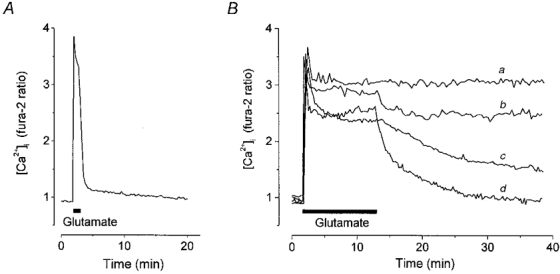
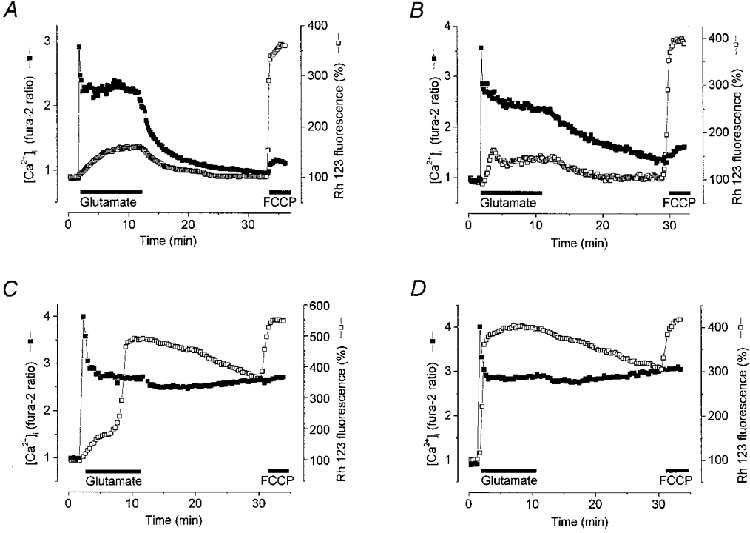
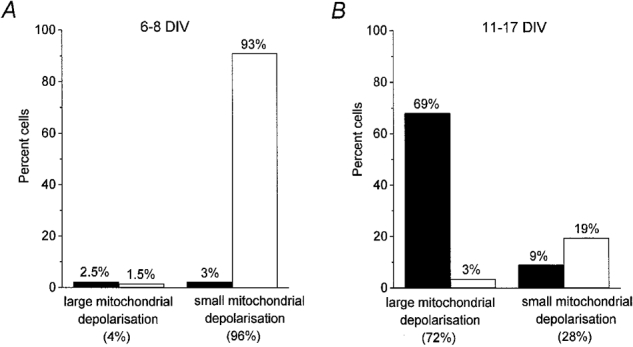
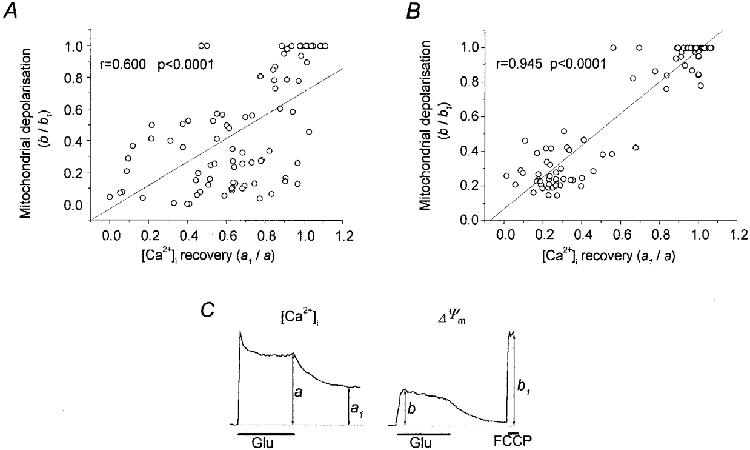
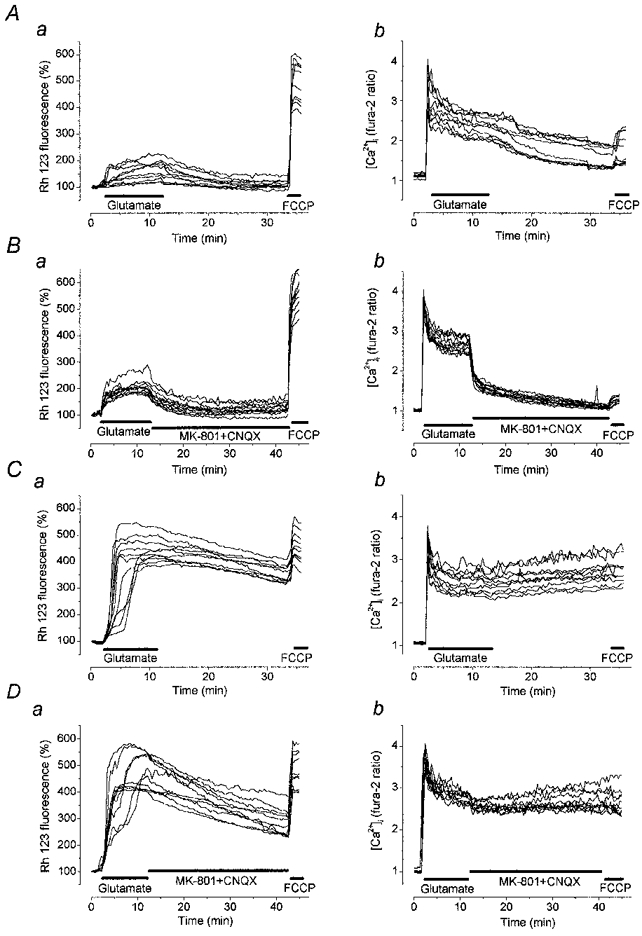
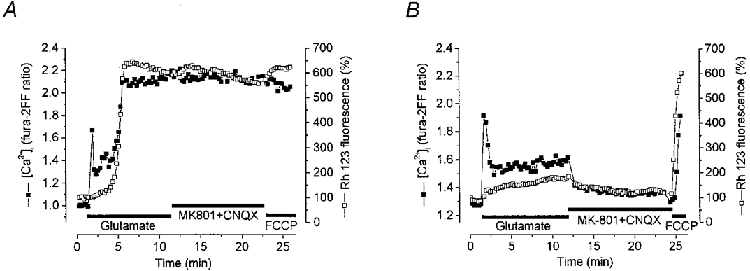
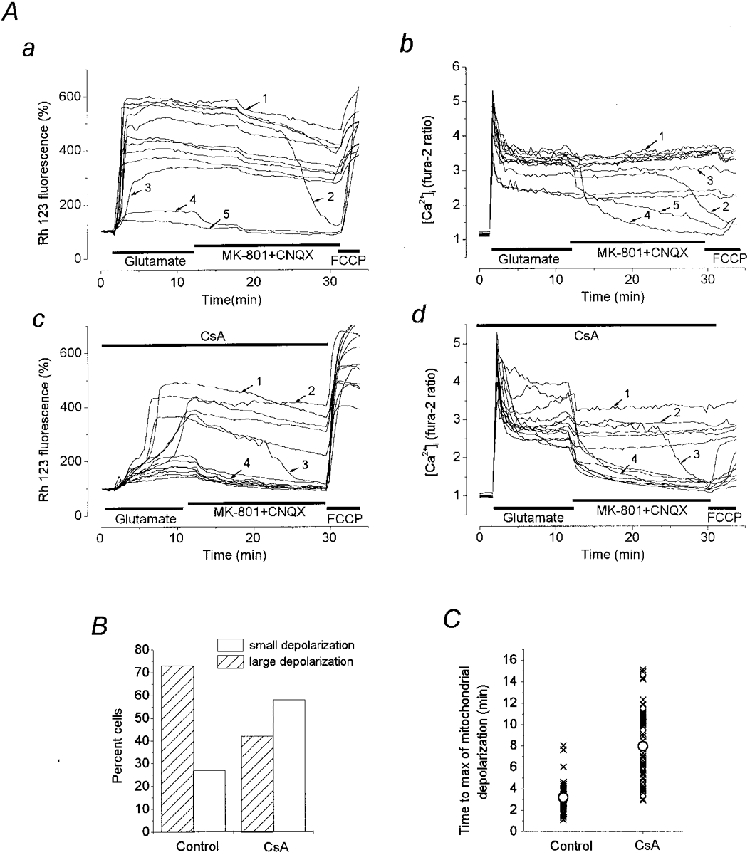
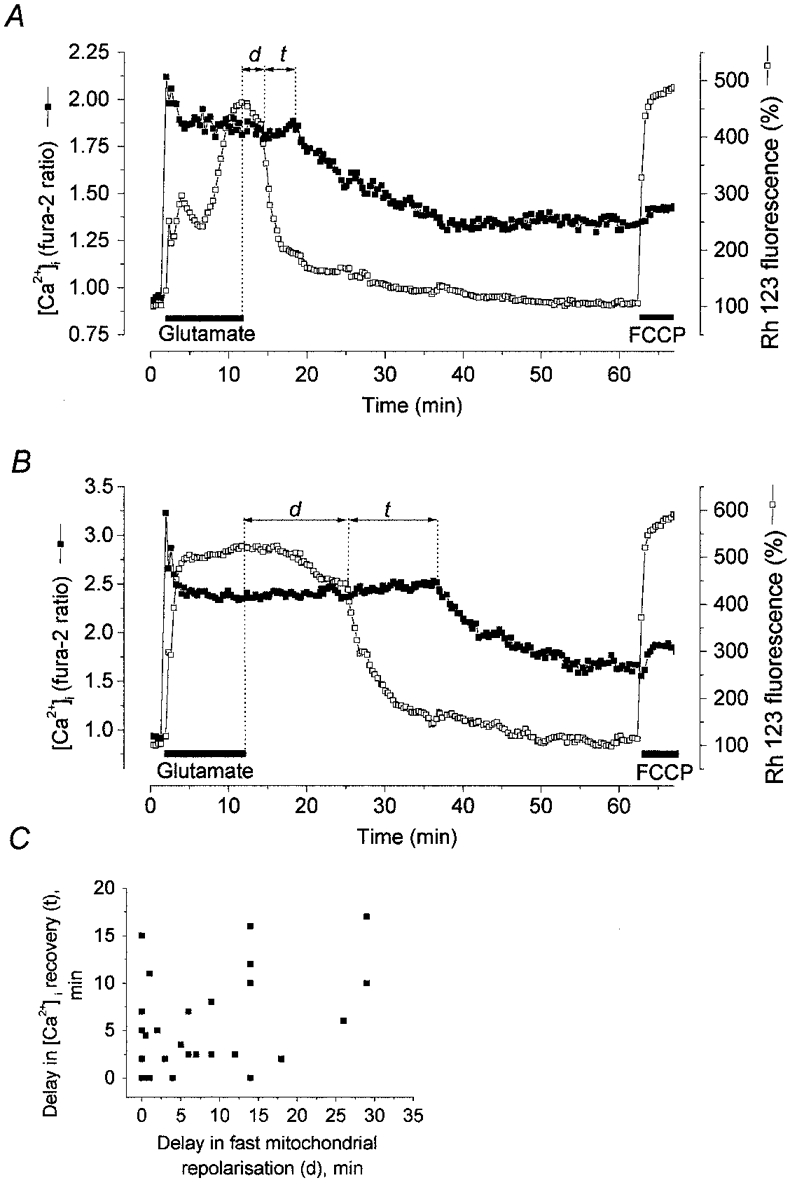
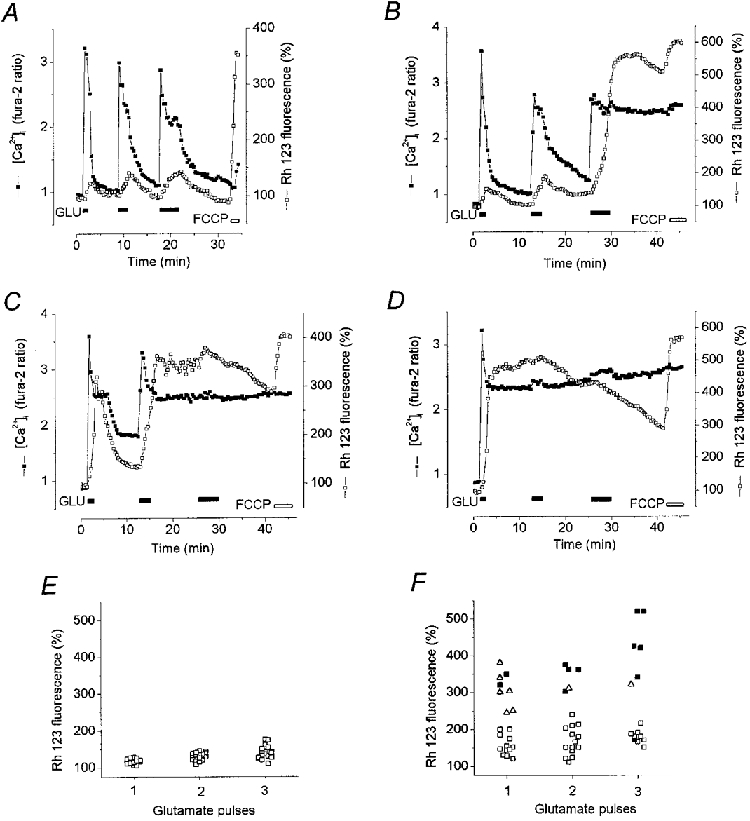
1. The objective of this study was to clarify the relationships between loss of mitochondrial potential and the perturbation of neuronal Ca2+ homeostasis induced by a toxic glutamate challenge. Digital fluorescence imaging techniques were employed to monitor simultaneously changes in cytoplasmic Ca2+ concentration ([Ca2+]i) and mitochondrial potential (DeltaPsim) in individual hippocampal neurones in culture coloaded with fura-2 AM or fura-2FF AM and rhodamine 123 (Rh 123). 2. In most cells (96 %) at 6-7 days in vitro (DIV) and in a small proportion of cells (29 %) at 11-17 DIV the [Ca2+]i increase induced by exposure to 100 microM glutamate for 10 min was associated with a small mitochondrial depolarisation, followed by mitochondrial repolarisation, and a degree of recovery of [Ca2+]i following glutamate washout. In the majority of neurones at 11-17 DIV (71 %), exposure to glutamate for 10 min induced a profound mono- or biphasic mitochondrial depolarisation, which was clearly correlated with a sustained [Ca2+]i plateau despite the removal of glutamate. 3. Addition of glutamate receptor antagonists (15 microM MK-801 plus 75 microM 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX)) to the washout solution did not affect the post-glutamate [Ca2+]i plateau in neurones exhibiting a profound mitochondrial depolarisation but greatly improved [Ca2+]i recovery in those neurones undergoing only a small mitochondrial depolarisation, suggesting that the release of endogenous glutamate delays [Ca2+]i recovery in the postglutamate period. 4. Cyclosporin A (500 nM) or N-methyl Val-4-cyclosporin A (200 nM) delayed or even prevented the development of the second phase of mitochondrial depolarisation in cells at 11-17 DIV and increased the proportion of neurones exhibiting a small monophasic mitochondrial depolarisation and [Ca2+]i recovery upon glutamate removal. 5. We have thus described a striking correlation between mitochondrial depolarisation and the failure of cells to restore [Ca2+]i following a toxic glutamate challenge. These data suggest that mitochondrial dysfunction plays a major role in the deregulation of [Ca2+]i associated with glutamate toxicity.
Figures
References
-
- Adamec E, Didier M, Nixon R. Developmental regulation of the recovery process following glutamate-induced calcium rise in rodent primary neuronal cultures. Developmental Brain Research. 1998;108:101–110. - PubMed
-
- Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Letters. 1996;394:321–324. - PubMed
-
- Augustine GJ, Neher E. Neuronal Ca2+ signalling takes the local route. Current Opinion in Neurobiology. 1992;2:302–307. - PubMed
-
- Bogachev A, Bykova L, Khodorov B, Andreeva N, Khaspekov L, Pinelis V, Victorov I. Stable decrease in ATP contents in cultured cerebellar and hippocampal neurons during and after toxic glutamate treatment. Biological Membranes. 1992;9:1057–1059. in Russian.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous