

Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia
- PMID: 10500204
- PMCID: PMC18061
- DOI: 10.1073/pnas.96.20.11492
Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia
Abstract
Friedreich ataxia (FRDA), the most common of the inherited ataxias, is an autosomal recessive degenerative disorder, characterized clinically by onset before the age of 25 of progressive gait and limb ataxia, absence of deep tendon reflexes, extensor plantar responses, and loss of position and vibration sense in the lower limbs. FRDA is caused by a GAA triplet expansion in the first intron of the FRDA gene on chromosome 9q13 in 97% of patients. The FRDA gene encodes a widely expressed 210-aa protein, frataxin, which is located in mitochondria and is severely reduced in FRDA patients. Frataxin function is still unknown but the knockout of the yeast frataxin homologue gene (YFH1) showed a severe defect of mitochondrial respiration and loss of mtDNA associated with elevated intramitochondrial iron. Here we report in vivo evidence of impaired mitochondrial respiration in skeletal muscle of FRDA patients. Using phosphorus magnetic resonance spectroscopy we demonstrated a maximum rate of muscle mitochondrial ATP production (V(max)) below the normal range in all 12 FRDA patients and a strong negative correlation between mitochondrial V(max) and the number of GAA repeats in the smaller allele. Our results show that FRDA is a nuclear-encoded mitochondrial disorder affecting oxidative phosphorylation and give a rationale for treatments aimed to improve mitochondrial function in this condition.
Figures
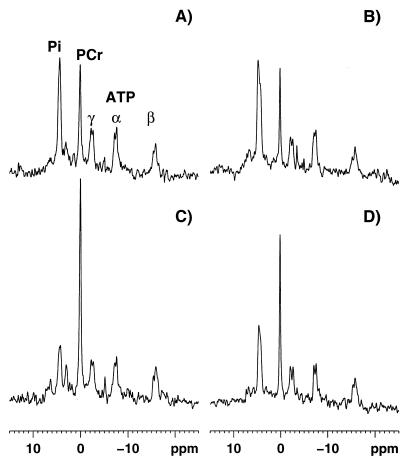
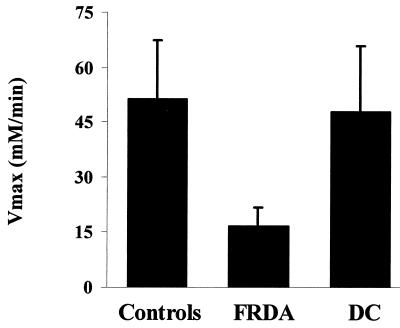
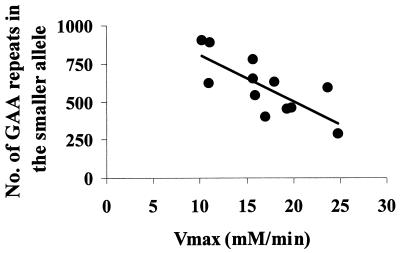
Comment in
-
Friedreich's ataxia is a mitochondrial disorder.Proc Natl Acad Sci U S A. 1999 Sep 28;96(20):10948-9. doi: 10.1073/pnas.96.20.10948. Proc Natl Acad Sci U S A. 1999. PMID: 10500103 Free PMC article. No abstract available.
References
-
- Harding A E. Brain. 1981;104:598–620. - PubMed
-
- Durr A, Cossee M, Agid Y, Capuzano V, Mignard C, Penet C, Mandel J L, Brice A, Koenig M. N Engl J Med. 1996;335:1169–1175. - PubMed
-
- Campuzano V, Montermini L, Moltò M D, Pianese L, Cassee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, et al. Science. 1996;271:1423–1426. - PubMed
-
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish S J, Faucheux B, Trouillas P, et al. Hum Mol Genet. 1997;6:1771–1780. - PubMed
-
- Babcock M, DeSilva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Science. 1997;276:1709–1712. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
