

The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events
- PMID: 10521296
- PMCID: PMC1288283
- DOI: 10.1086/302623
The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events
Abstract
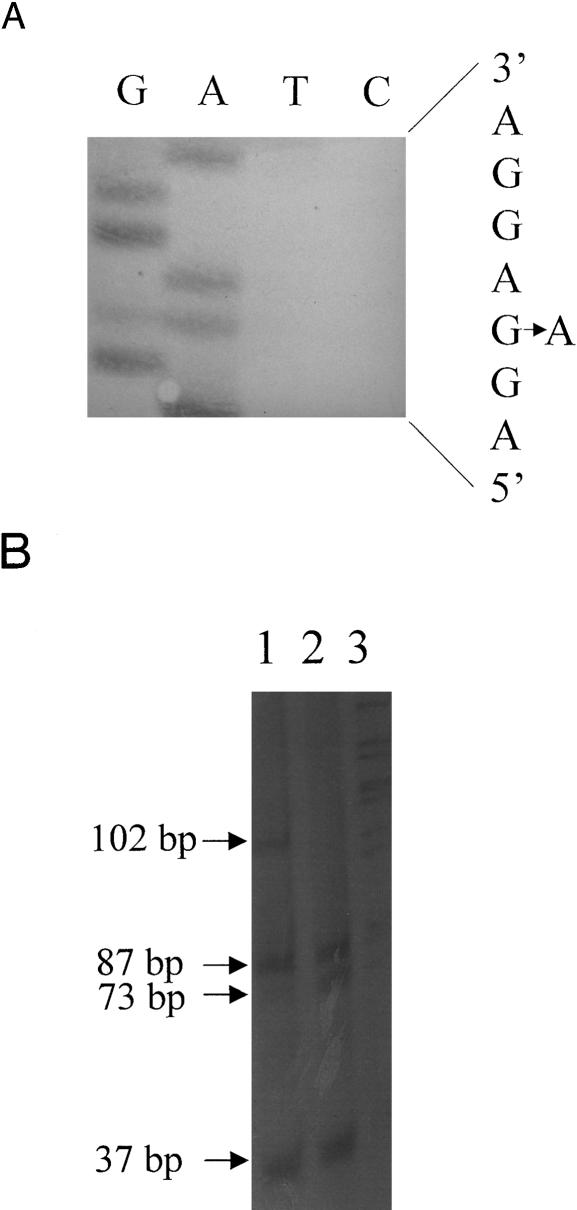
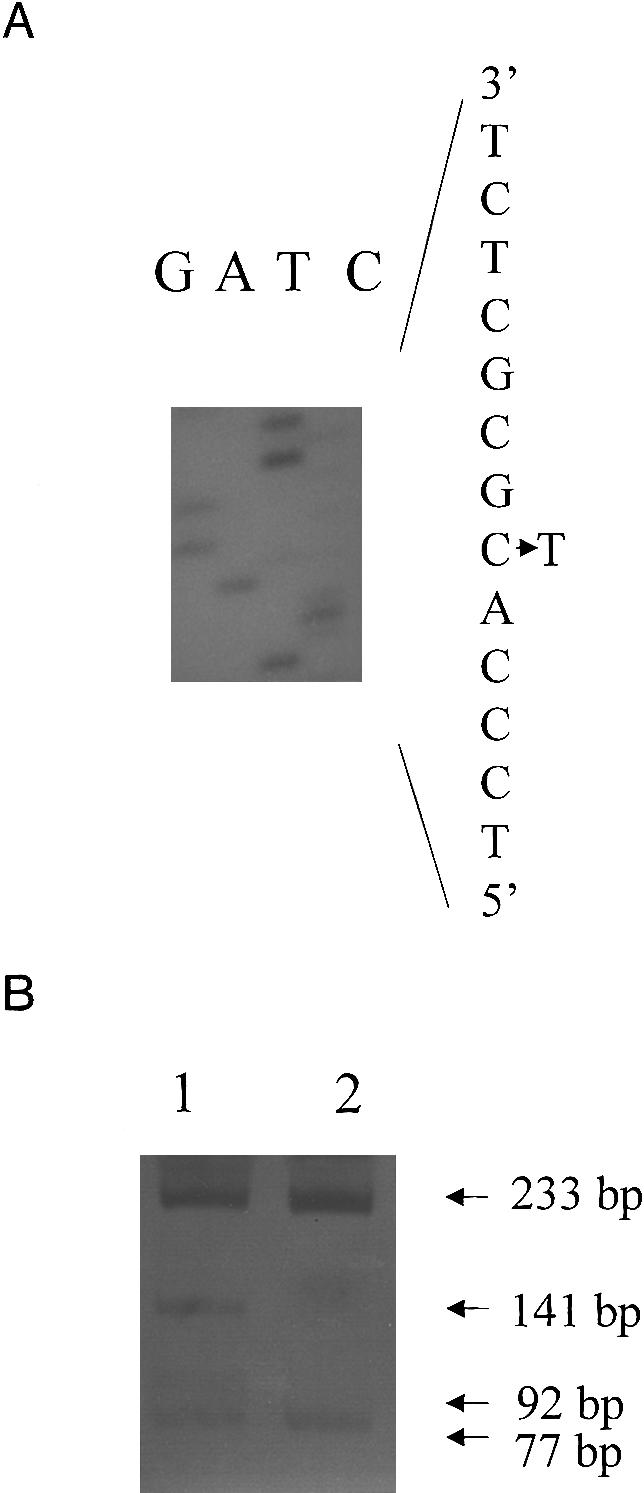
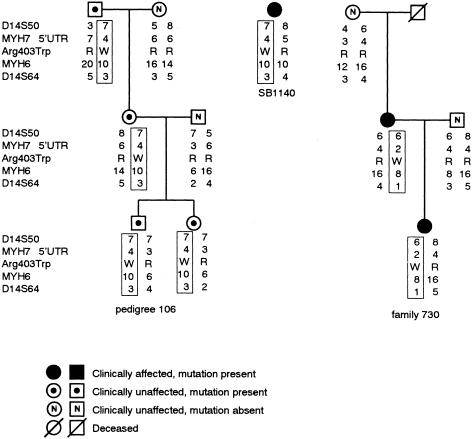
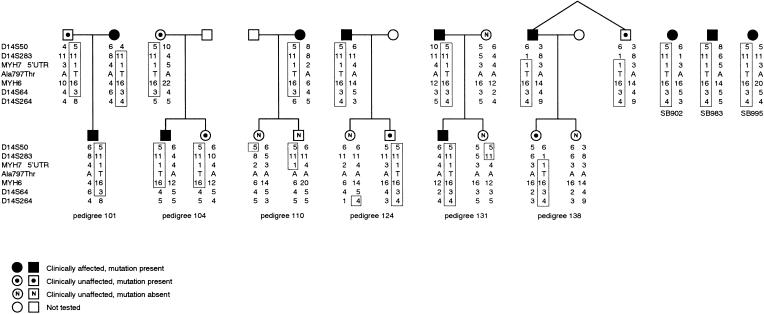
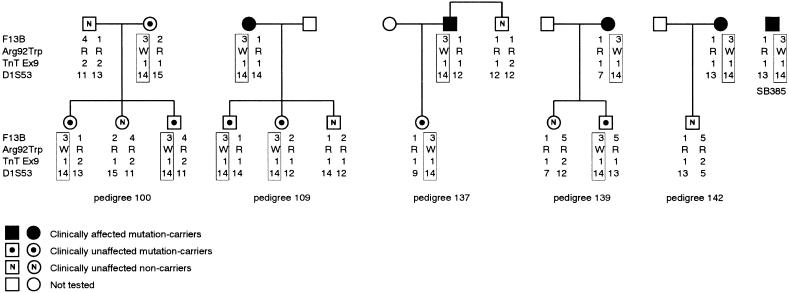
Hypertrophic cardiomyopathy (HCM) is an autosomal dominantly inherited disease of the cardiac sarcomere, caused by numerous mutations in genes encoding protein components of this structure. Mutation carriers are at risk of sudden cardiac death, mostly as adolescents or young adults. The reproductive disadvantage incurred may explain both the global occurrence of diverse independent HCM-associated mutations and the rare reports of founder effects within populations. We have investigated whether this holds true for two South African subpopulations, one of mixed ancestry and one of northern-European descent. Previously, we had detected three novel mutations-Ala797Thr in the beta-myosin heavy-chain gene (betaMHC), Arg92Trp in the cardiac troponin T gene (cTnT), and Arg645His in the myosin-binding protein C gene (MyBPC)-and two documented betaMHC mutations (Arg403Trp and Arg249Gln). Here we report three additional novel mutations-Gln499Lys in betaMHC and Val896Met and Deltac756 in MyBPC-and the documented betaMHC Arg719Gln mutation. Seven of the nine HCM-causing mutations arose independently; no conclusions can be drawn for the remaining two. However, the betaMHC Arg403Trp and Ala797Thr and cTnT Arg92Trp mutations were detected in another one, eight, and four probands, respectively, and haplotype analysis in families carrying these recurring mutations inferred their origin from three common ancestors. The milder phenotype of the betaMHC mutations may account for the presence of these founder effects, whereas population dynamics alone may have overridden the reproductive disadvantage incurred by the more lethal, cTnT Arg92Trp mutation.
Figures

References
Electronic-Database Information
-
- DNA Mutation Database, http://www.angis.org.au/Databases/Heart/heartbreak.html (for familial HCM)
-
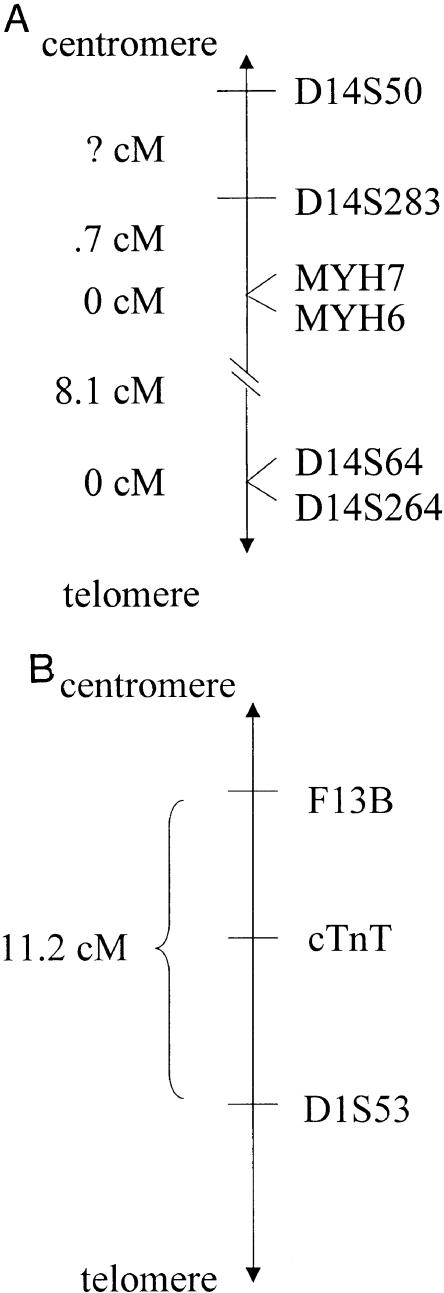
- GeneMap '99, http://www.ncbi.nlm.nih.gov/genemap/map.cgi?CHR=14
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim (for HCM [MIM 192600])
References
-
- Bonne G, Carrier L, Richard P, Hainque B, Schwartz K (1998) Familial hypertrophic cardiomyopathy from mutations to functional defects. Circ Res 83:580–593 - PubMed
-
- Botha MC, Beighton P (1983) Inherited disorders in the Afrikaner population of southern Africa. I. Historical and demographic background, cardiovascular, neurological, metabolic and intestinal conditions. S Afr Med J 64:609–612 - PubMed
-
- Botha MC, Pritchard J (1972) Blood group frequencies: an indication of the genetic constitution of population samples in Cape Town. S Afr Med J Suppl 46:1–27 - PubMed
-
- Brink PA, Ferreira A, Moolman JC, Weymar HW, Van der Merwe P-L, Corfield VA (1995) Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation 91:1633–1640 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
