

Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis
- PMID: 10521396
- PMCID: PMC317073
- DOI: 10.1101/gad.13.19.2514
Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis
Abstract
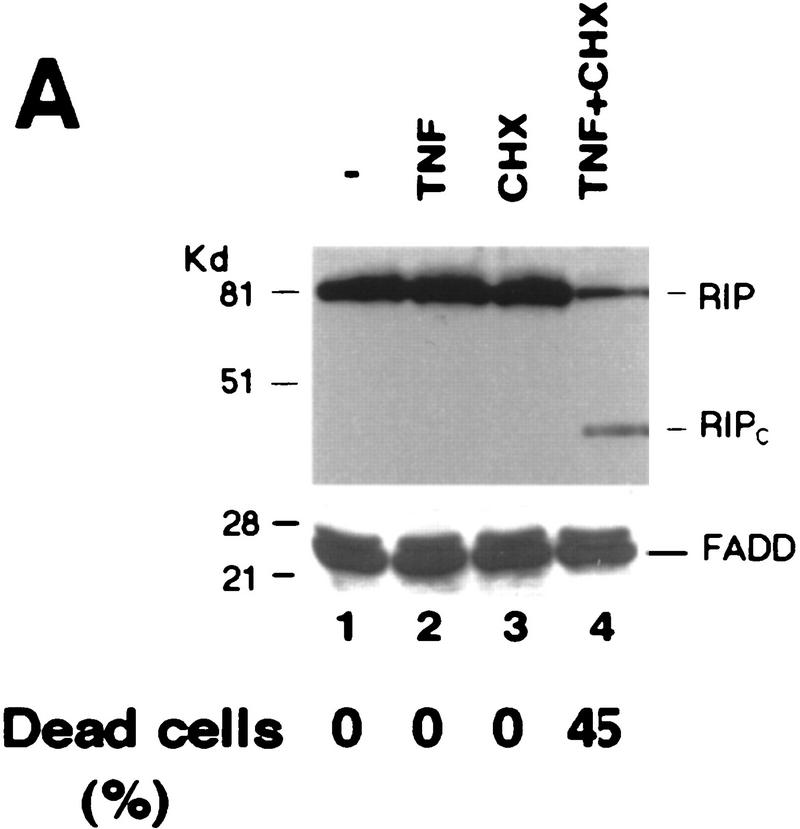
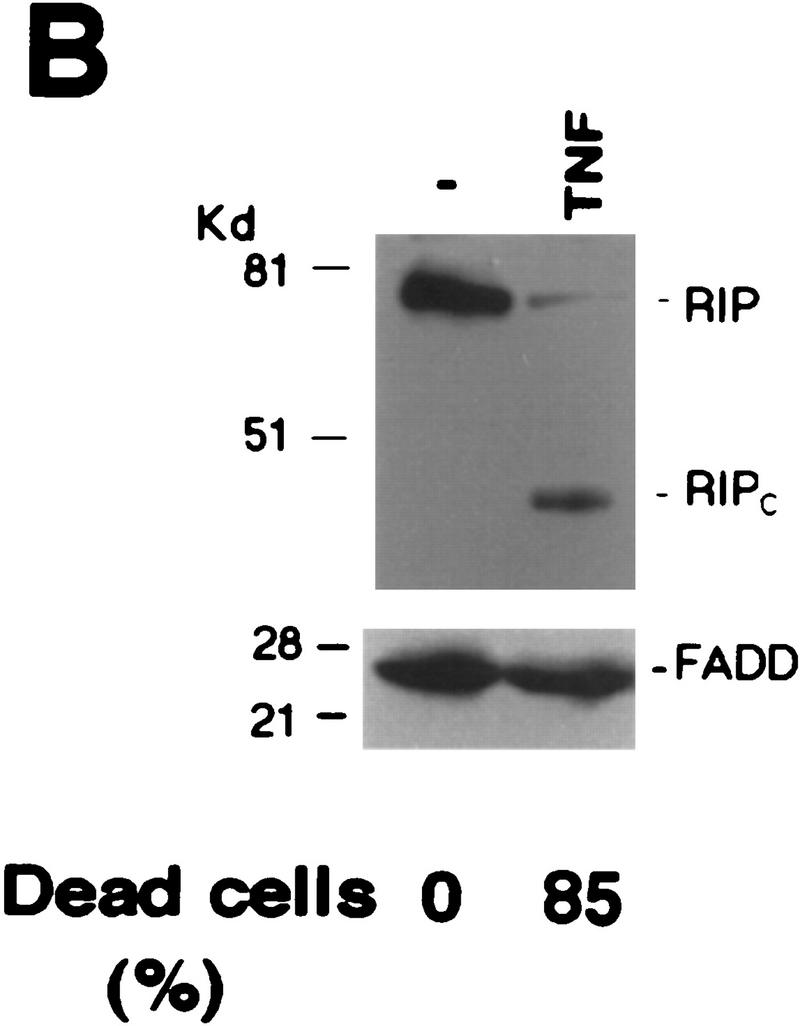
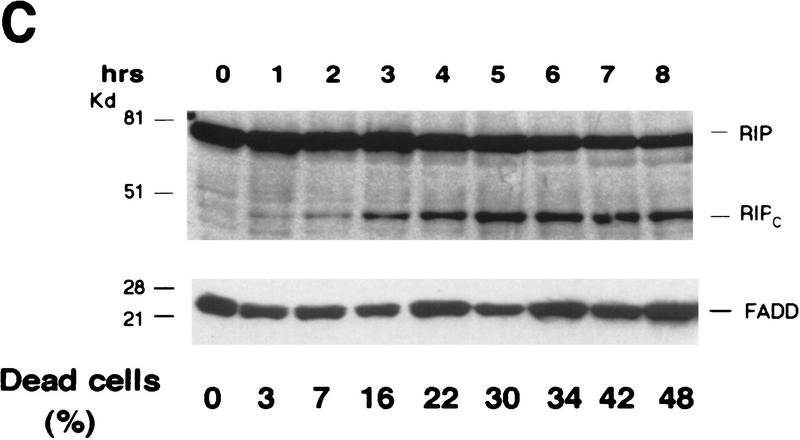
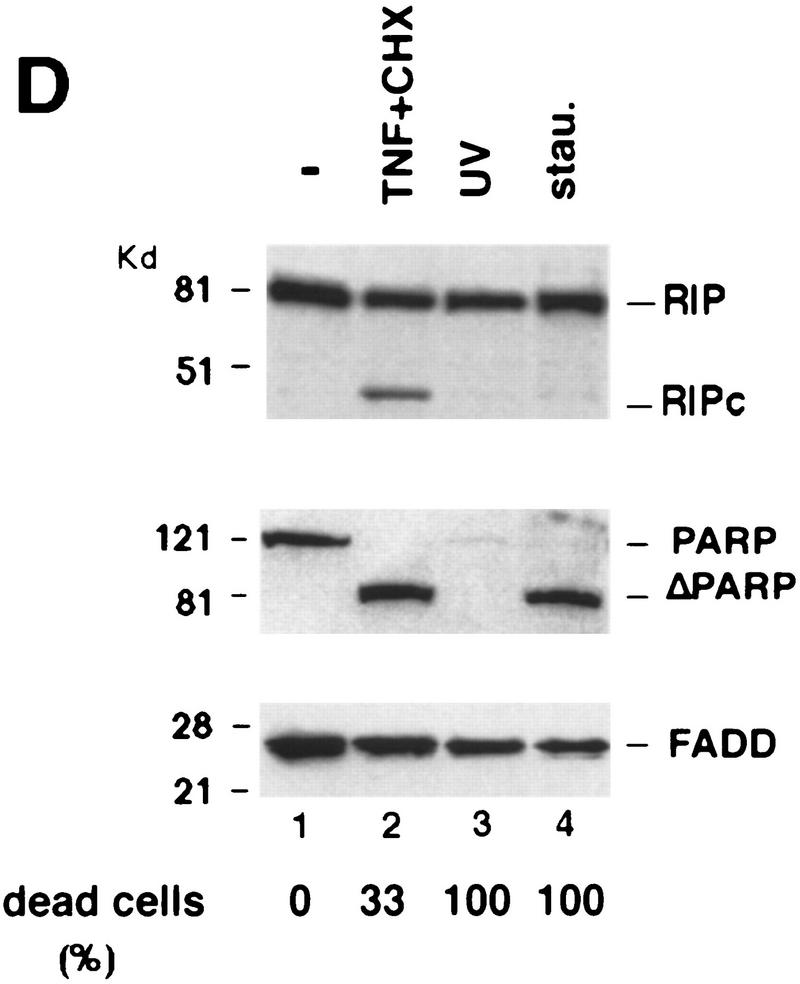
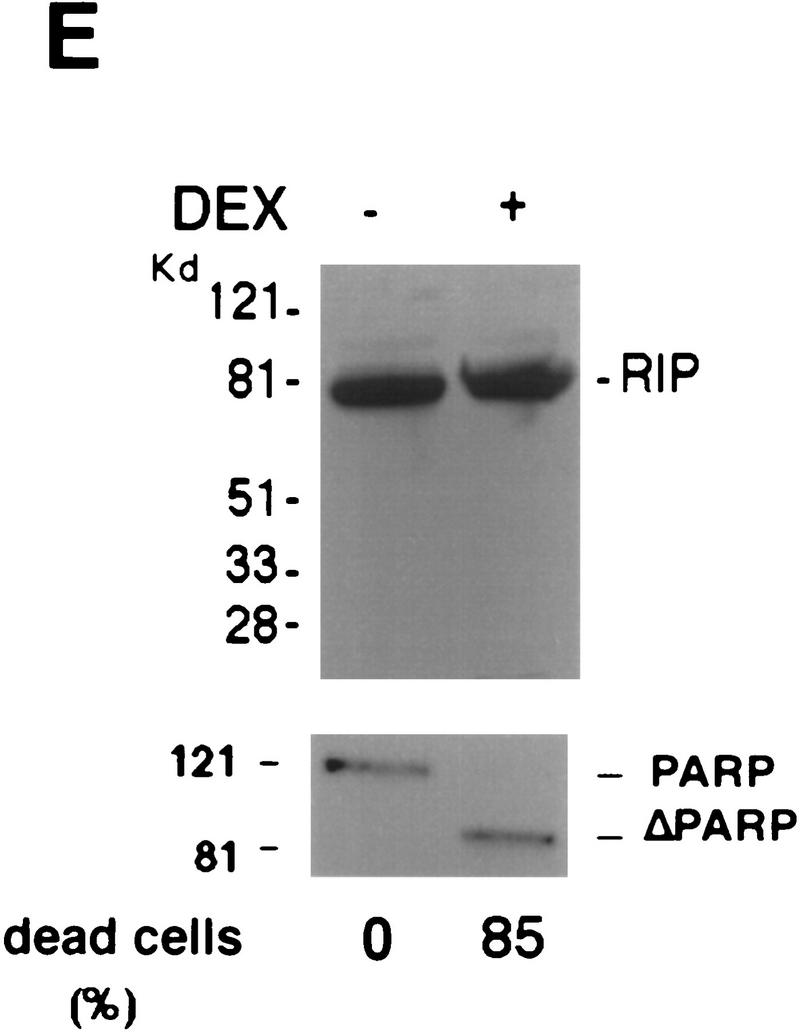
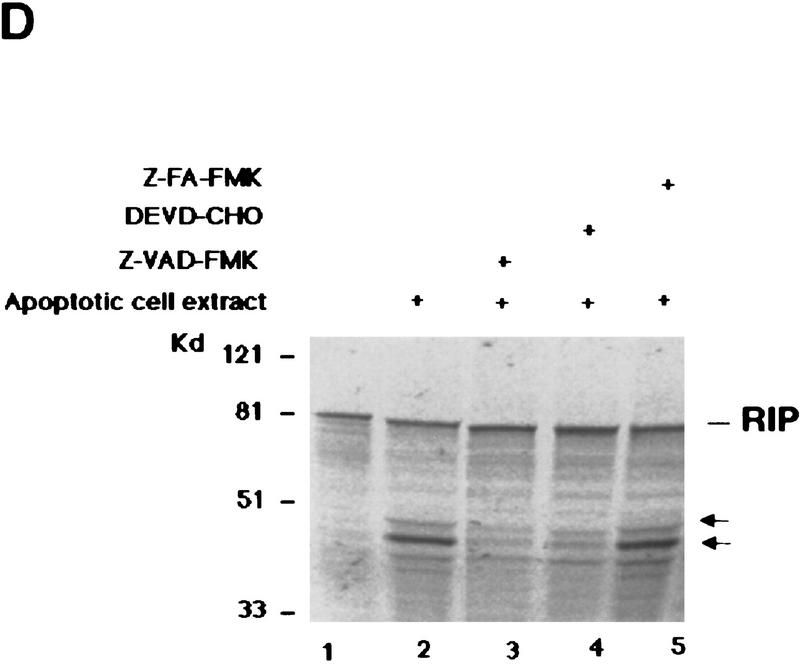
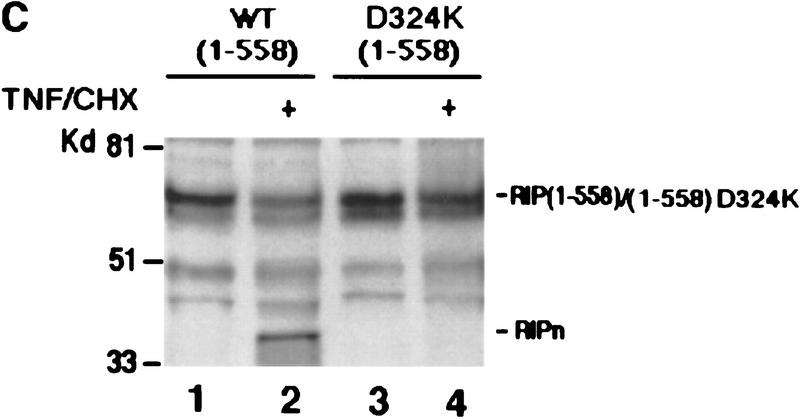
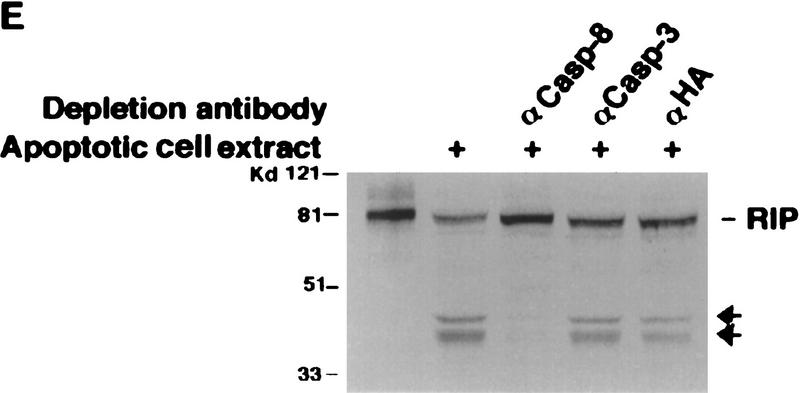
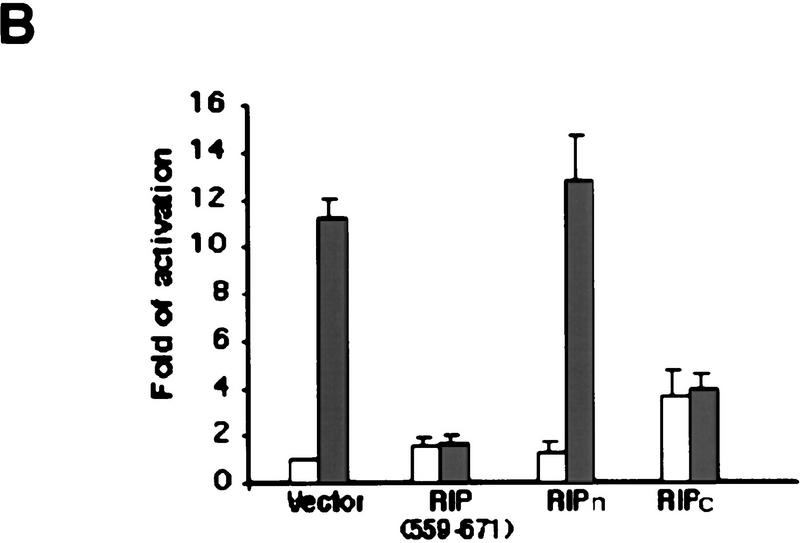
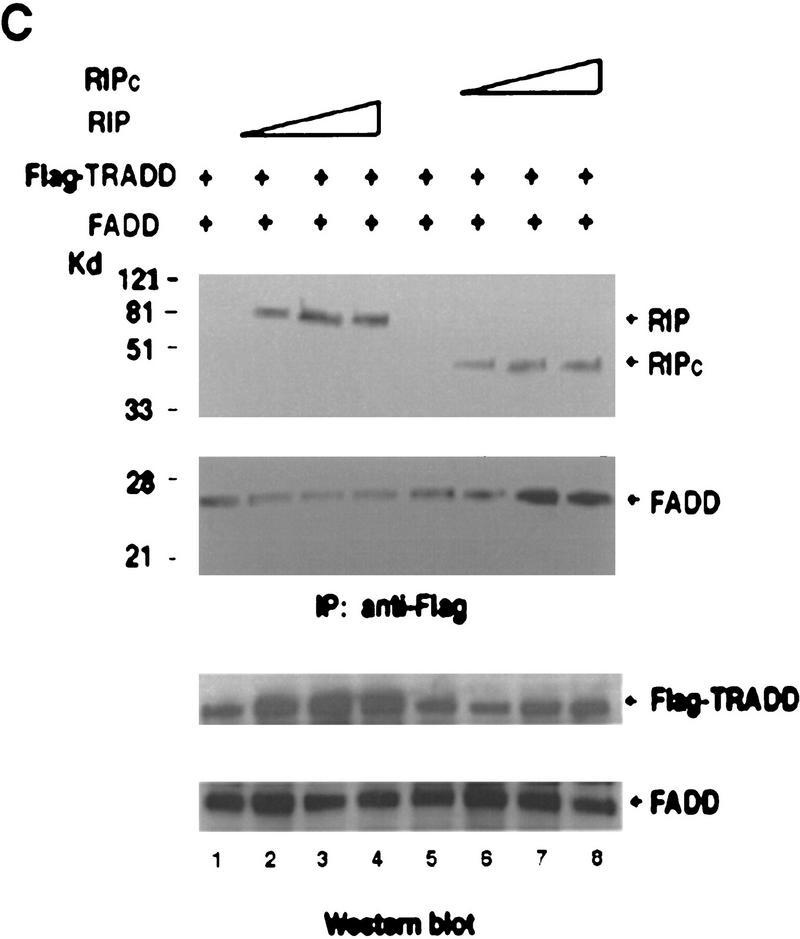
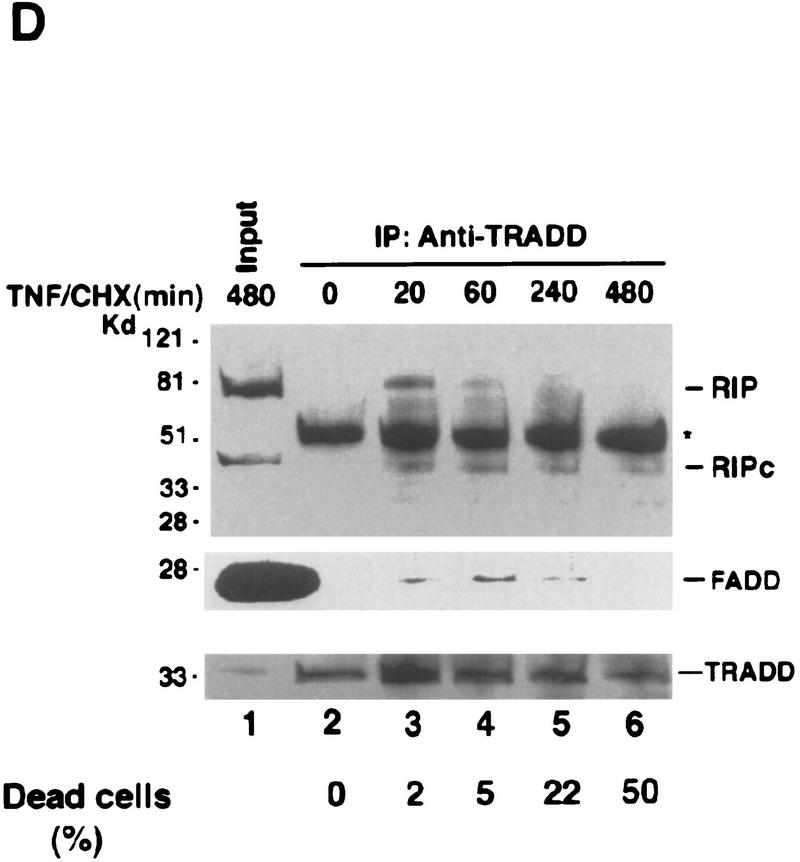
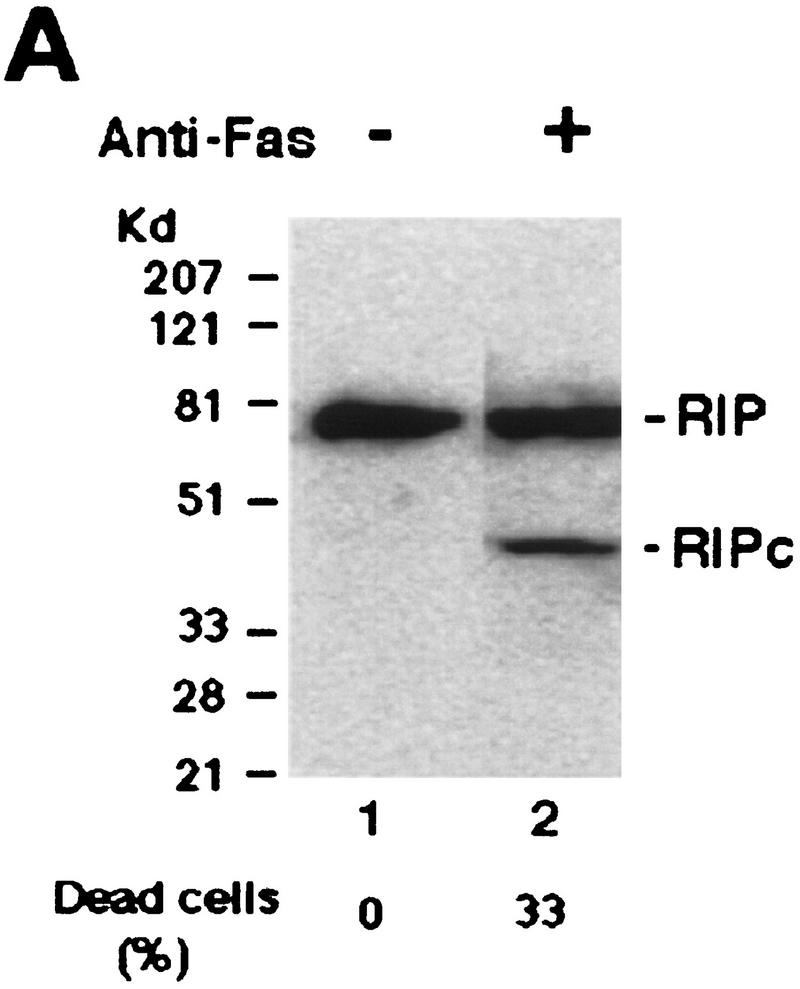
Although the molecular mechanisms of TNF signaling have been largely elucidated, the principle that regulates the balance of life and death is still unknown. We report here that the death domain kinase RIP, a key component of the TNF signaling complex, was cleaved by Caspase-8 in TNF-induced apoptosis. The cleavage site was mapped to the aspartic acid at position 324 of RIP. We demonstrated that the cleavage of RIP resulted in the blockage of TNF-induced NF-kappaB activation. RIPc, one of the cleavage products, enhanced interaction between TRADD and FADD/MORT1 and increased cells' sensitivity to TNF. Most importantly, the Caspase-8 resistant RIP mutants protected cells against TNF-induced apopotosis. These results suggest that cleavage of RIP is an important process in TNF-induced apoptosis. Further more, RIP cleavage was also detected in other death receptor-mediated apoptosis. Therefore, our study provides a potential mechanism to convert cells from life to death in death receptor-mediated apoptosis.
Figures











References
-
- Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. 1998;281:1305–1308. - PubMed
-
- Baeuerle PA. IκB-NF-κB structures: At the interface of inflammation control. Cell. 1998;95:729–731. - PubMed
-
- Baeuerle PA, Baltimore D. NF-κB: Ten years after. Cell. 1996;87:13–20. - PubMed
-
- Baker SJ, Reddy EP. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261–3270. - PubMed
-
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNFα-induced cell death. Science. 1996;274:782–784. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous