

Steroid disorders in children: congenital adrenal hyperplasia and apparent mineralocorticoid excess
- PMID: 10536001
- PMCID: PMC23101
- DOI: 10.1073/pnas.96.22.12790
Steroid disorders in children: congenital adrenal hyperplasia and apparent mineralocorticoid excess
Abstract
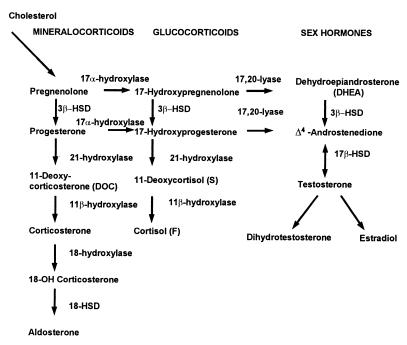
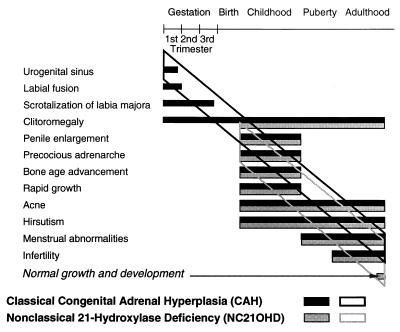
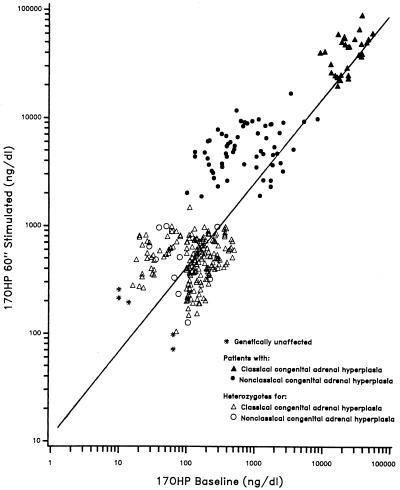
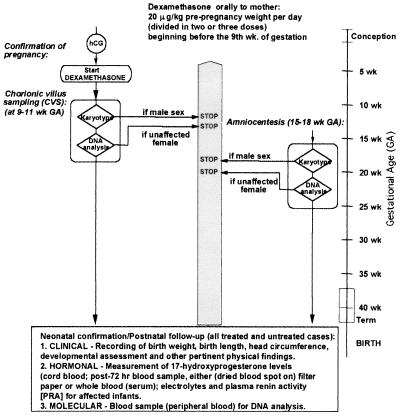
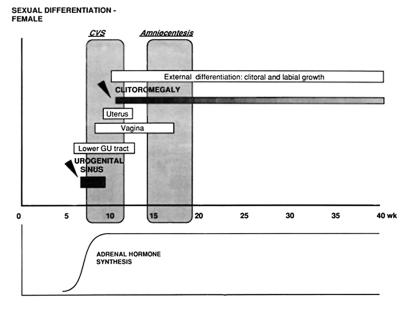
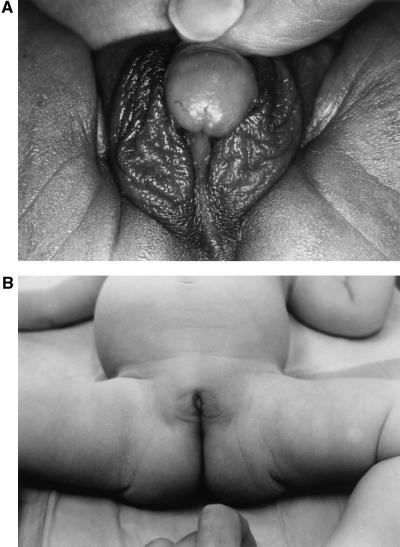
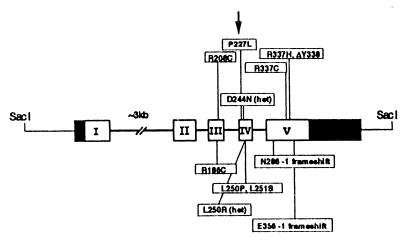
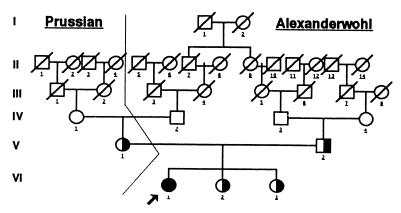
Our research team and laboratories have concentrated on two inherited endocrine disorders, congenital adrenal hyperplasia (CAH) and apparent mineralocorticoid excess, in thier investigations of the pathophysiology of adrenal steroid hormone disorders in children. CAH refers to a family of inherited disorders in which defects occur in one of the enzymatic steps required to synthesize cortisol from cholesterol in the adrenal gland. Because of the impaired cortisol secretion, adrenocorticotropic hormone levels rise due to impairment of a negative feedback system, which results in hyperplasia of the adrenal cortex. The majority of cases is due to 21-hydroxylase deficiency (21-OHD). Owing to the blocked enzymatic step, cortisol precursors accumulate in excess and are converted to potent androgens, which are secreted and cause in utero virilization of the affected female fetus genitalia in the classical form of CAH. A mild form of the 21-OHD, termed nonclassical 21-OHD, is the most common autosomal recessive disorder in humans, and occurs in 1/27 Ashkenazic Jews. Mutations in the CYP21 gene have been identified that cause both classical and nonclassical CAH. Apparent mineralocorticoid excess is a potentially fatal genetic disorder causing severe juvenile hypertension, pre- and postnatal growth failure, and low to undetectable levels of potassium, renin, and aldosterone. It is caused by autosomal recessive mutations in the HSD11B2 gene, which result in a deficiency of 11beta-hydroxysteroid dehydrogenase type 2. In 1998, we reported a mild form of this disease, which may represent an important cause of low-renin hypertension.
Figures
References
-
- De Crecchio L. Morgagni. 1865;7:154–188.
-
- New M I, Dupont B, Grumbach K, Levine L S. In: The Metabolic Basis of Inherited Disease. Stanbury J B, Wyngaarden J B, Fredrickson D S, Goldstein J L, Brown M D, editors. New York: McGraw–Hill; 1982. pp. 973–1000.
-
- Lin D, Sugawara T, Strauss J F, 3rd, Clark B J, Stocco D M, Saenger P, Rogol A, Miller W L. Science. 1995;267:1821–1831. - PubMed
-
- New M I, White P C. In: Genetic and Molecular Biological Aspects of Endocrine Disease. Thakker R V, editor. London: Bailliere Tindall; 1995. pp. 525–554.
-
- Jost A. In: Hermaphroditism, Genital Anomalies and Related Endocrine Disorders. Jones H W, Scott W W, editors. Baltimore: Williams & Wilkins; 1971. p. 16.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
