

INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53
- PMID: 10541553
- PMCID: PMC317110
- DOI: 10.1101/gad.13.20.2670
INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53
Abstract
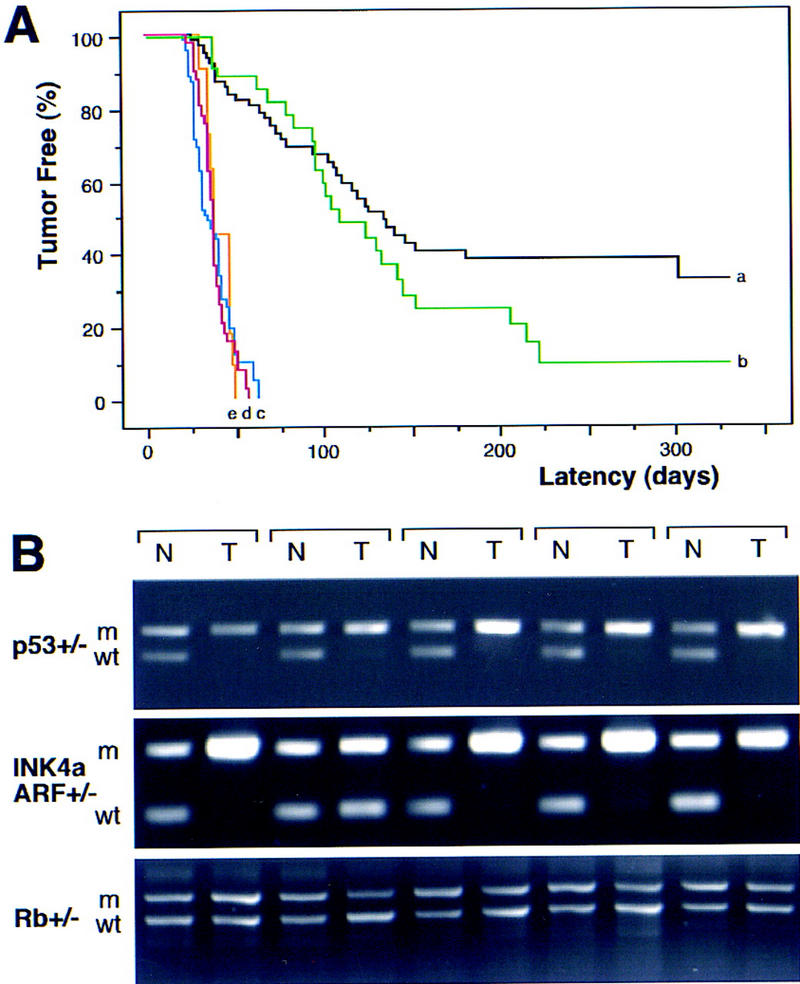
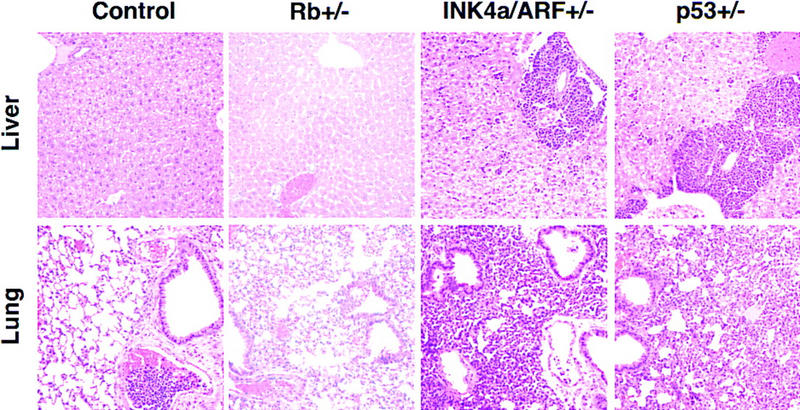
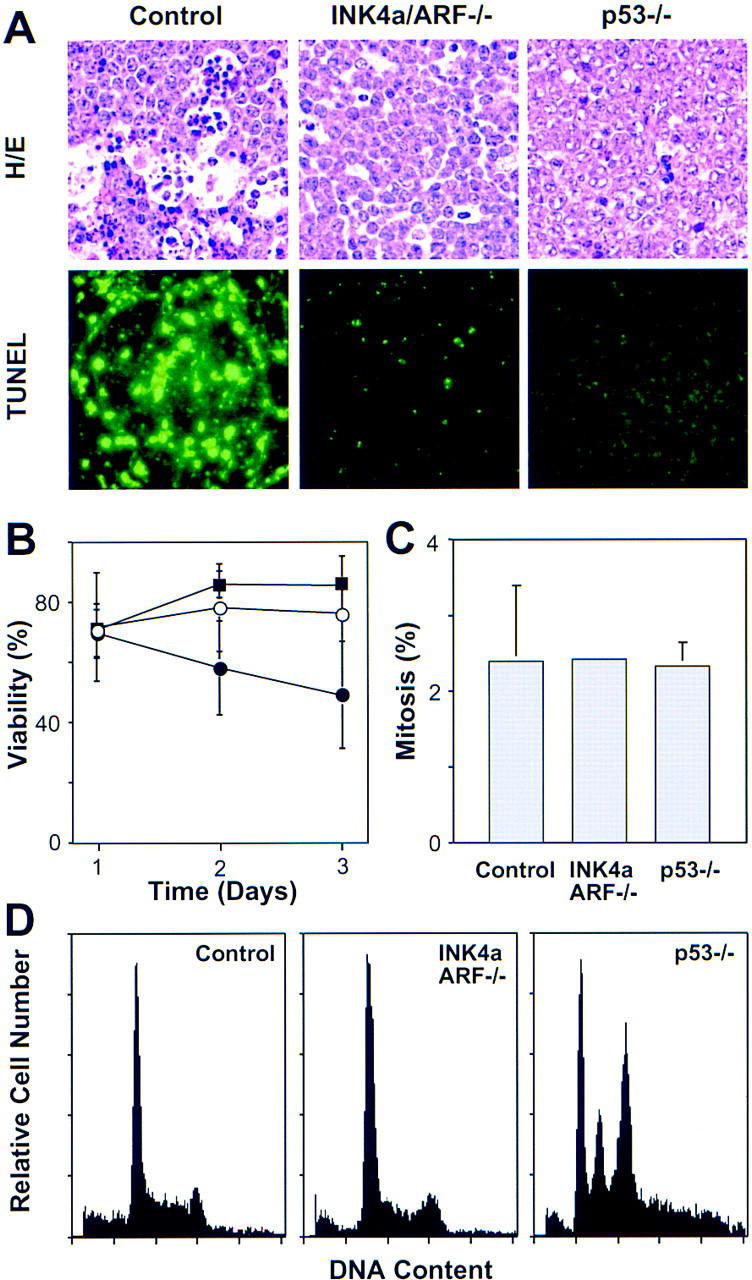
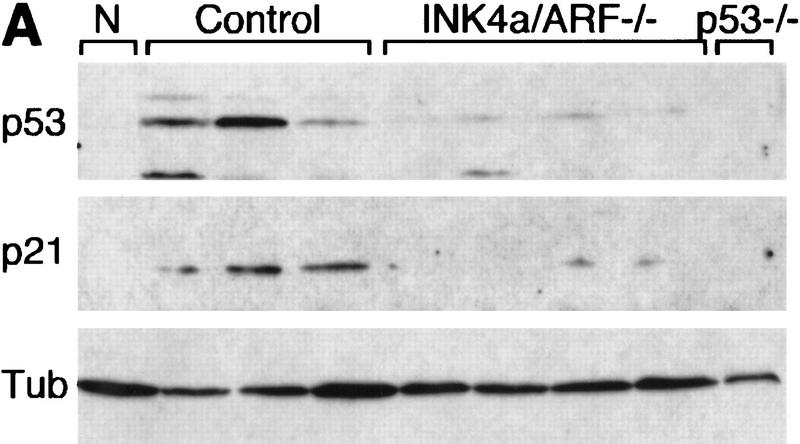
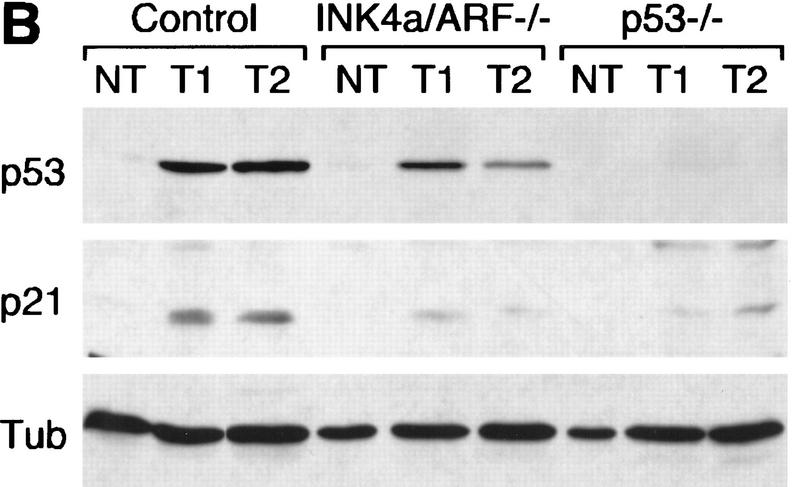
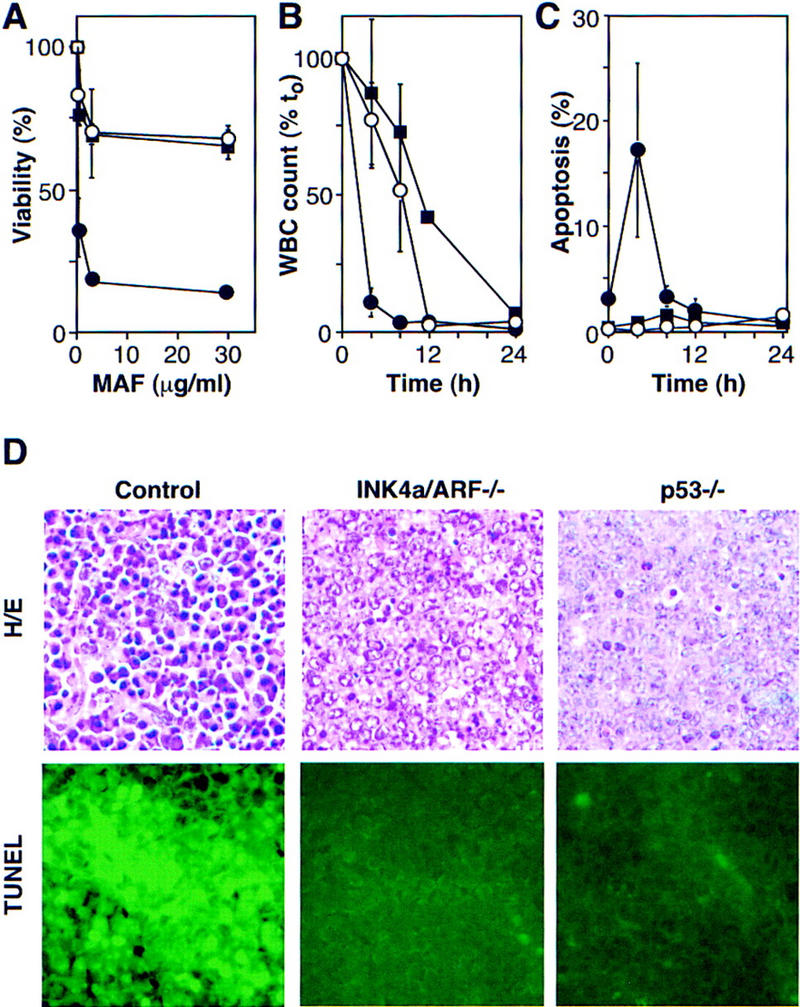
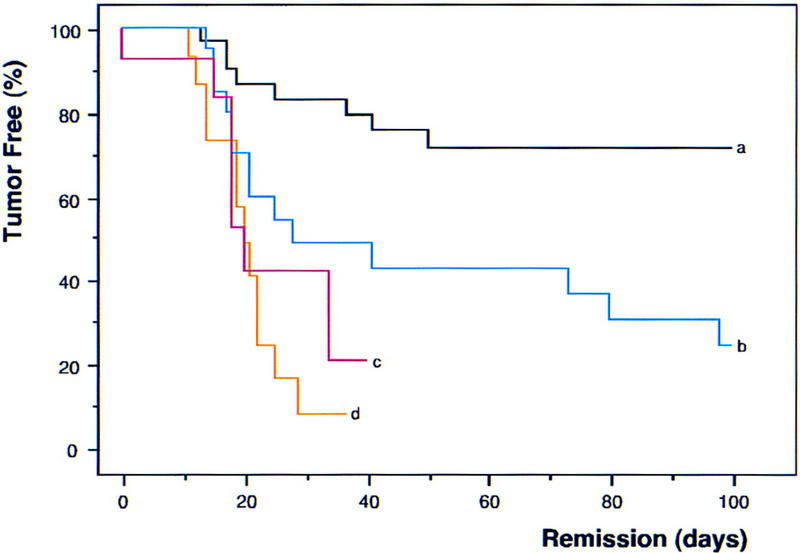
The INK4a/ARF locus encodes upstream regulators of the retinoblastoma and p53 tumor suppressor gene products. To compare the impact of these loci on tumor development and treatment response, the Emu-myc transgenic lymphoma model was used to generate genetically defined tumors with mutations in the INK4a/ARF, Rb, or p53 genes. Like p53 null lymphomas, INK4a/ARF null lymphomas formed rapidly, were highly invasive, displayed apoptotic defects, and were markedly resistant to chemotherapy in vitro and in vivo. Furthermore, INK4a/ARF(-/-) lymphomas displayed reduced p53 activity despite the presence of wild-type p53 genes. Consequently, INK4a/ARF and p53 mutations lead to aggressive tumors by disrupting overlapping tumor suppressor functions. These data have important implications for understanding the clinical behavior of human tumors.
Figures
References
-
- Adams JM, Cory S. Transgenic models for haemopoietic malignancies. Biochim Biophys Acta. 1991;1072:9–31. - PubMed
-
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. - PubMed
-
- Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–125. - PubMed
-
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999;59:1391–1399. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous