

Mismatch repair processing of carcinogen-DNA adducts triggers apoptosis
- PMID: 10567554
- PMCID: PMC84913
- DOI: 10.1128/MCB.19.12.8292
Mismatch repair processing of carcinogen-DNA adducts triggers apoptosis
Abstract
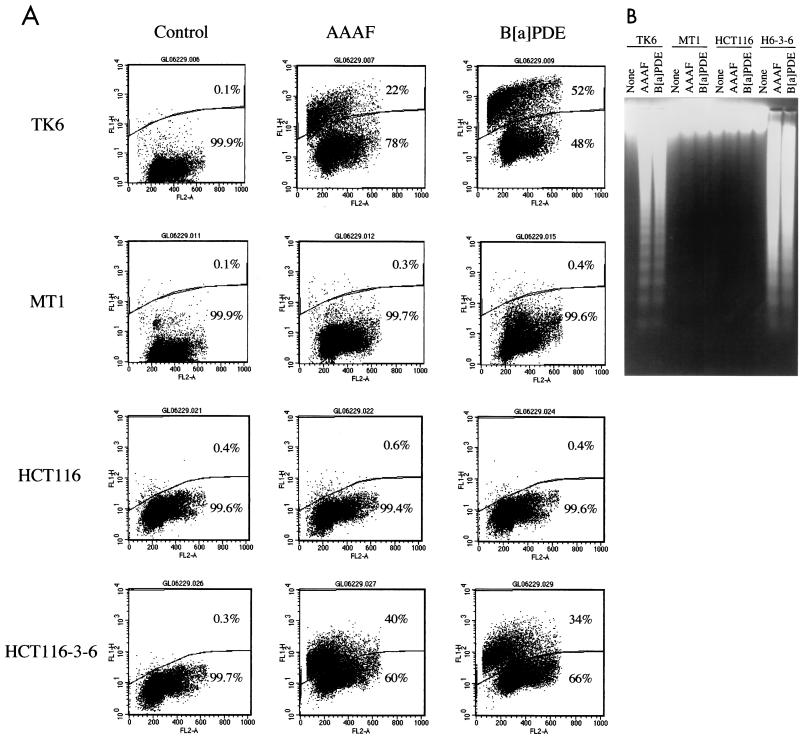
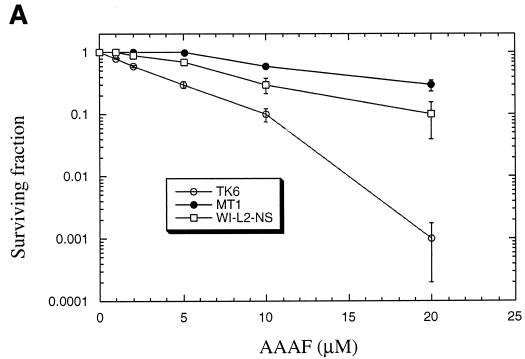
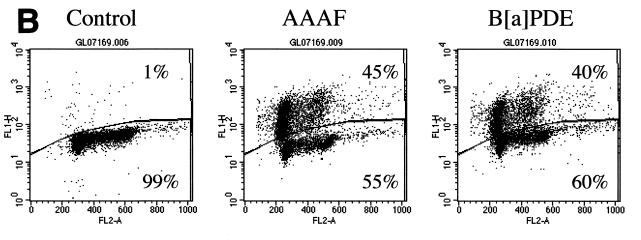
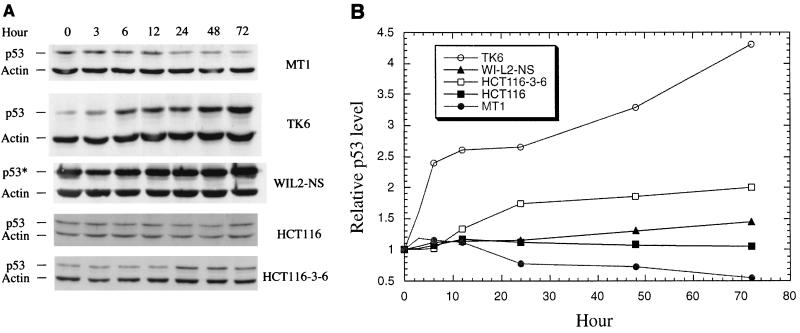
The DNA mismatch repair pathway is well known for its role in correcting biosynthetic errors of DNA replication. We report here a novel role for mismatch repair in signaling programmed cell death in response to DNA damage induced by chemical carcinogens. Cells proficient in mismatch repair were highly sensitive to the cytotoxic effects of chemical carcinogens, while cells defective in either human MutS or MutL homologs were relatively insensitive. Since wild-type cells but not mutant cells underwent apoptosis upon treatment with chemical carcinogens, the apoptotic response is dependent on a functional mismatch repair system. By analyzing p53 expression in several pairs of cell lines, we found that the mismatch repair-dependent apoptotic response was mediated through both p53-dependent and p53-independent pathways. In vitro biochemical studies demonstrated that the human mismatch recognition proteins hMutSalpha and hMutSbeta efficiently recognized DNA damage induced by chemical carcinogens, suggesting a direct participation of mismatch repair proteins in mediating the apoptotic response. Taken together, these studies further elucidate the mechanism by which mismatch repair deficiency predisposes to cancer, i.e., the deficiency not only causes a failure to repair mismatches generated during DNA metabolism but also fails to direct damaged and mutation-prone cells to commit suicide.
Figures
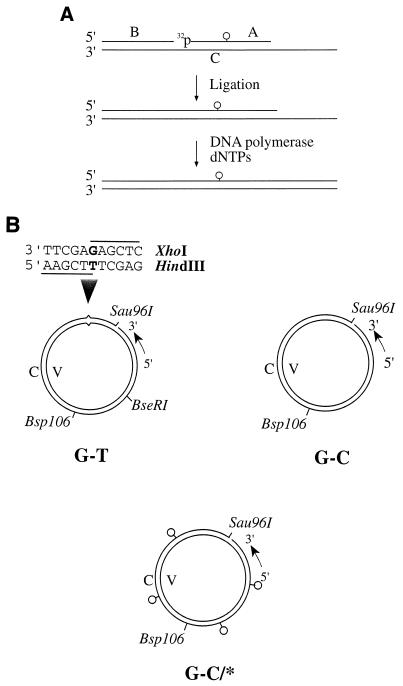
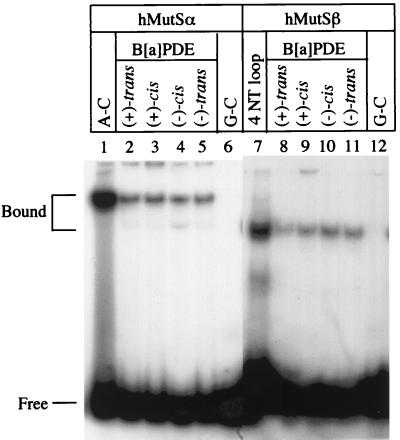
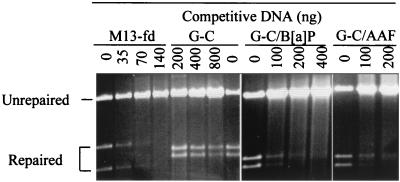
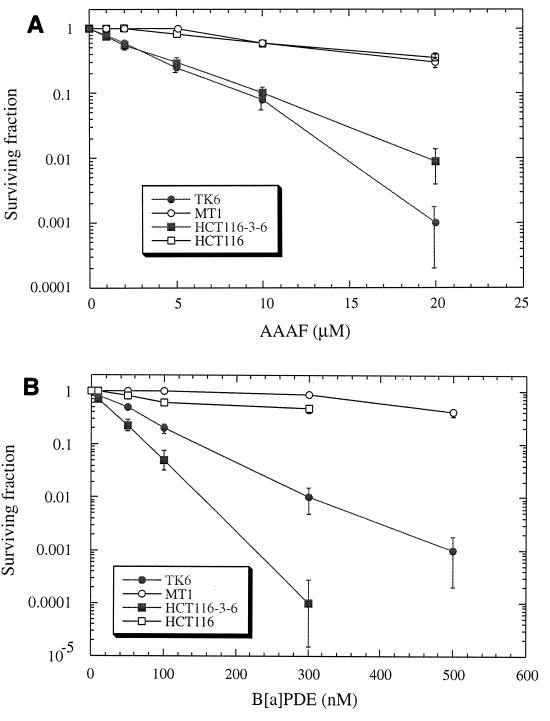

References
-
- Alani E, Lee S, Kane M F, Griffith J, Kolodner R D. Saccharomyces cerevisiae MSH2, a mispaired base recognition protein, also recognizes Holliday junctions in DNA. J Mol Biol. 1997;265:289–301. - PubMed
-
- Anthoney D A, McIlwrath A J, Gallagher W M, Edlin A R M, Brown R. Microsatellite instability, apoptosis, and loss of p53 function in drug-resistant tumor cells. Cancer Res. 1996;56:1374–1381. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous