

Bacterial diversity within the human subgingival crevice
- PMID: 10588742
- PMCID: PMC24473
- DOI: 10.1073/pnas.96.25.14547
Bacterial diversity within the human subgingival crevice
Abstract
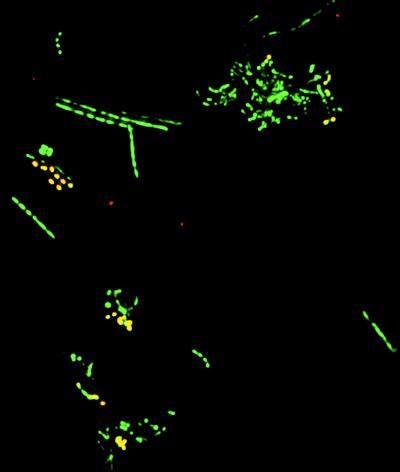
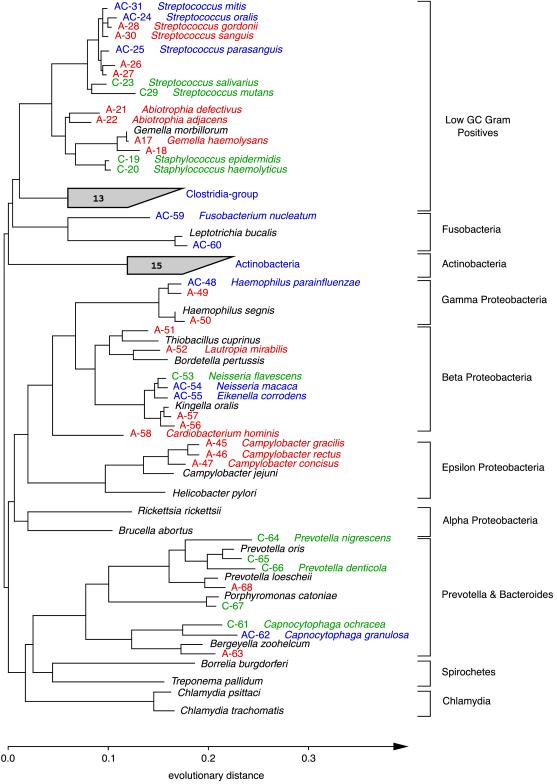
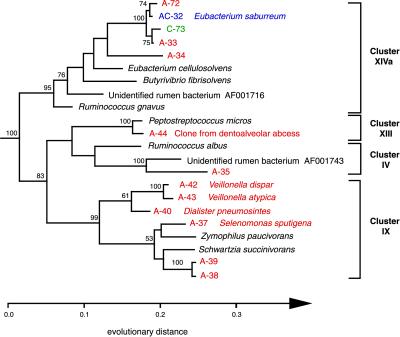
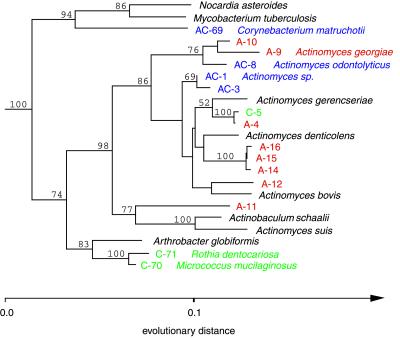
Molecular, sequence-based environmental surveys of microorganisms have revealed a large degree of previously uncharacterized diversity. However, nearly all studies of the human endogenous bacterial flora have relied on cultivation and biochemical characterization of the resident organisms. We used molecular methods to characterize the breadth of bacterial diversity within the human subgingival crevice by comparing 264 small subunit rDNA sequences from 21 clone libraries created with products amplified directly from subgingival plaque, with sequences obtained from bacteria that were cultivated from the same specimen, as well as with sequences available in public databases. The majority (52.5%) of the directly amplified 16S rRNA sequences were <99% identical to sequences within public databases. In contrast, only 21.4% of the sequences recovered from cultivated bacteria showed this degree of variability. The 16S rDNA sequences recovered by direct amplification were also more deeply divergent; 13.5% of the amplified sequences were more than 5% nonidentical to any known sequence, a level of dissimilarity that is often found between members of different genera. None of the cultivated sequences exhibited this degree of sequence dissimilarity. Finally, direct amplification of 16S rDNA yielded a more diverse view of the subgingival bacterial flora than did cultivation. Our data suggest that a significant proportion of the resident human bacterial flora remain poorly characterized, even within this well studied and familiar microbial environment.
Figures
References
-
- Mackowiak P A. N Engl J Med. 1982;307:83–93. - PubMed
-
- Giovannoni S J, Britschgi T B, Moyer C L, Field K G. Nature (London) 1990;345:60–63. - PubMed
-
- Stackebrandt E, Liesack W, Goebel B M. FASEB J. 1993;7:232–236. - PubMed
-
- Relman D A, Loutit J S, Schmidt T M, Falkow S, Tompkins L S. N Engl J Med. 1990;323:1573–1580. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
