

Correlation between point mutations in NS2 and the viability and cytopathogenicity of Bovine viral diarrhea virus strain Oregon analyzed with an infectious cDNA clone
- PMID: 10590128
- PMCID: PMC111550
- DOI: 10.1128/jvi.74.1.390-400.2000
Correlation between point mutations in NS2 and the viability and cytopathogenicity of Bovine viral diarrhea virus strain Oregon analyzed with an infectious cDNA clone
Abstract
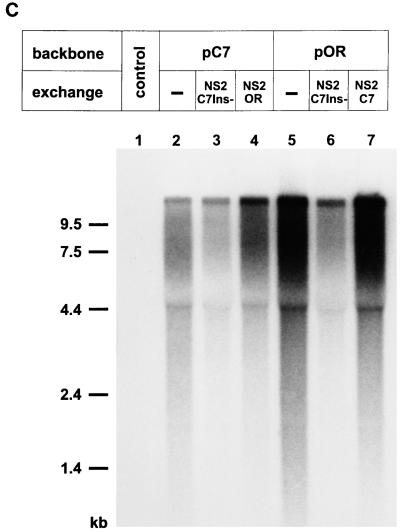
Cytopathogenicity of Bovine viral diarrhea virus (BVDV) is correlated with expression of the nonstructural protein NS3, which can be generated by processing of a fusion protein termed NS2-3. For the cytopathogenic (cp) BVDV strain Oregon, NS2-3 processing is based on a set of point mutations within NS2. To analyze the correlation between NS2-3 cleavage and cytopathogenicity, a full-length cDNA clone composed of cDNA from BVDV Oregon and the utmost 5'- and 3'-terminal sequences of a published infectious BVDV clone was established. After transfection of RNA transcribed from this cDNA clone, infectious virus with similar growth characteristics to wild-type BVDV Oregon could be recovered that also exhibited a cytopathic effect. Based on this cDNA construct and published cp and noncp infectious clones, chimeric full-length cDNA clones were constructed. Analysis of the recovered viruses demonstrated that the presence of the NS2 gene of BVDV Oregon in a chimeric construct is sufficient for NS2-3 processing and a cp phenotype. Since previous studies had revealed that the amino acid serine at position 1555 of BVDV Oregon plays an important role in efficient NS2-3 cleavage, mutants of BVDV Oregon with different amino acids at this position were constructed. Some of these mutants showed NS2-3 cleavage efficiencies in the range of the wild-type sequence and allowed the recovery of viruses that behaved similarly to wild-type virus with regard to growth characteristics and cytopathogenicity. In contrast, other mutants with considerably reduced NS2-3 cleavage efficiencies propagated much more slowly and reverted to viruses expressing polyproteins with sequences allowing efficient NS2-3 cleavage. These viruses apparently induced cytopathic effects only after reversion.
Figures

References
-
- Baker J C. Bovine viral diarrhea virus: a review. J Am Vet Med Assoc. 1987;1990:1449–1458. - PubMed
-
- Becher P, Shannon A D, Tautz N, Thiel H-J. Molecular characterization of border disease virus, a pestivirus from sheep. Virology. 1994;198:542–551. - PubMed
-
- Bolin S R, McClurkin A W, Cutlip R C, Coria M F. Severe clinical disease induced in cattle persistently infected with noncytopathic bovine viral diarrhea virus by superinfection with cytopathic bovine viral diarrhea virus. Am J Vet Res. 1985;46:573–576. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
