

Biological characteristics of the leukemia-associated transcriptional factor AML1 disclosed by hematopoietic rescue of AML1-deficient embryonic stem cells by using a knock-in strategy
- PMID: 10594034
- PMCID: PMC85087
- DOI: 10.1128/MCB.20.1.319-328.2000
Biological characteristics of the leukemia-associated transcriptional factor AML1 disclosed by hematopoietic rescue of AML1-deficient embryonic stem cells by using a knock-in strategy
Abstract
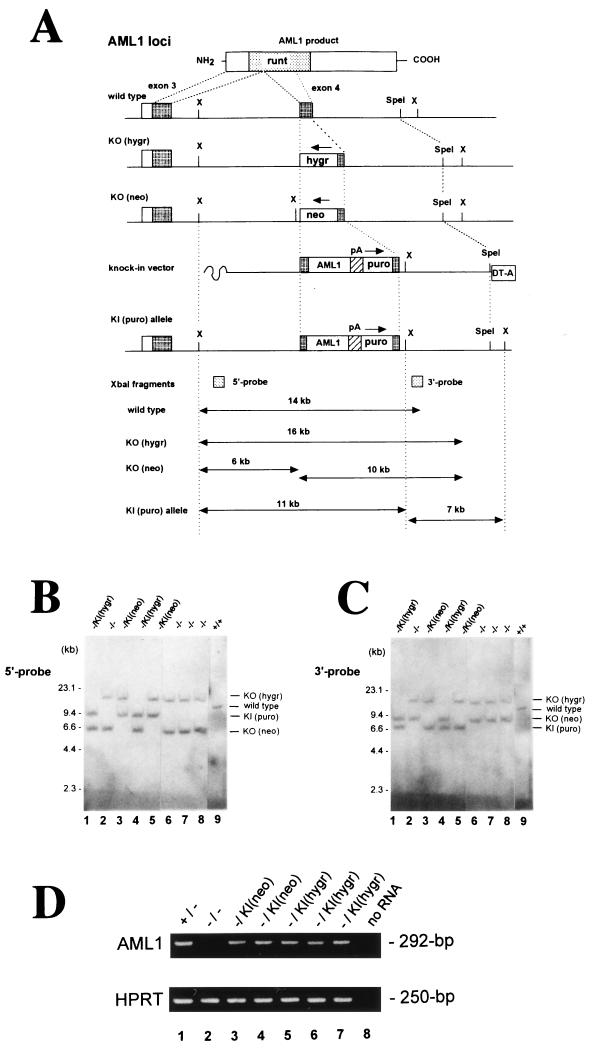
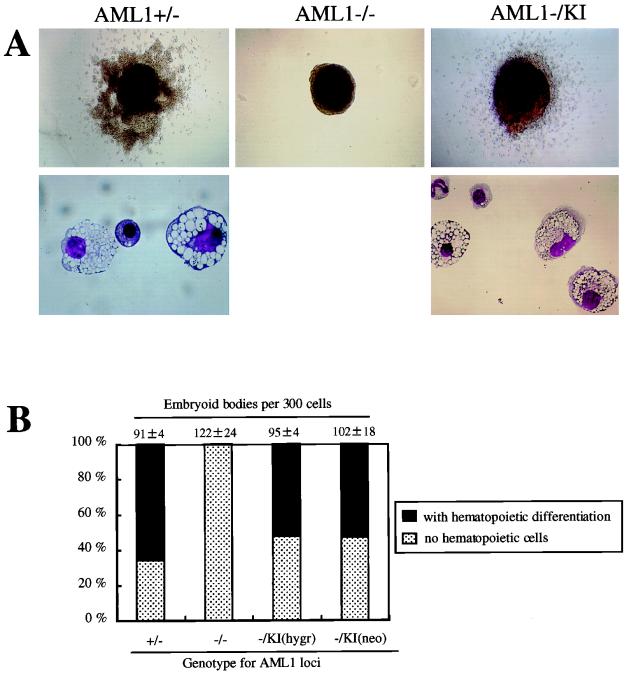
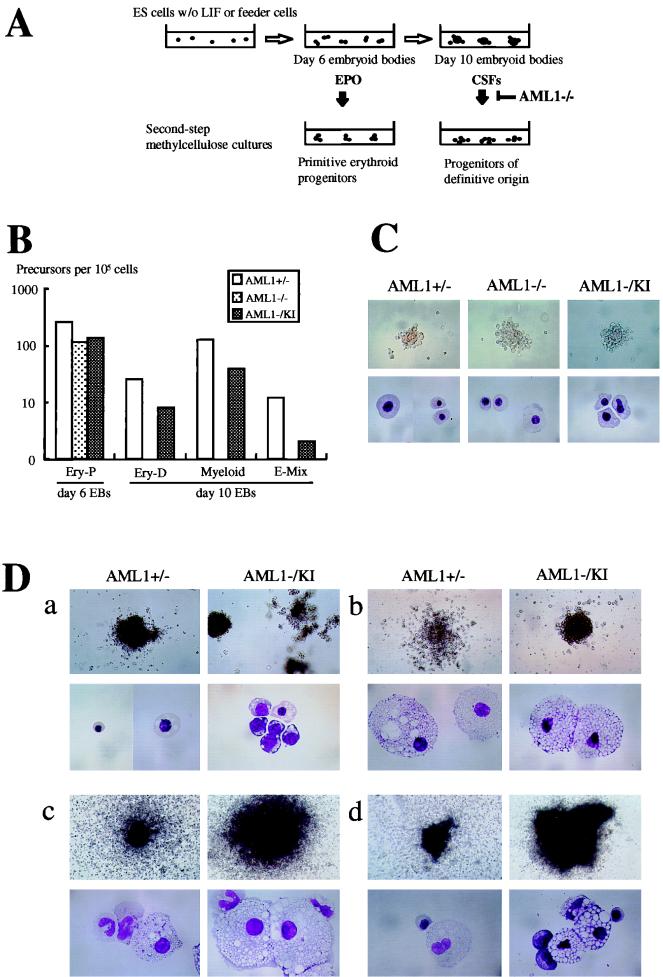
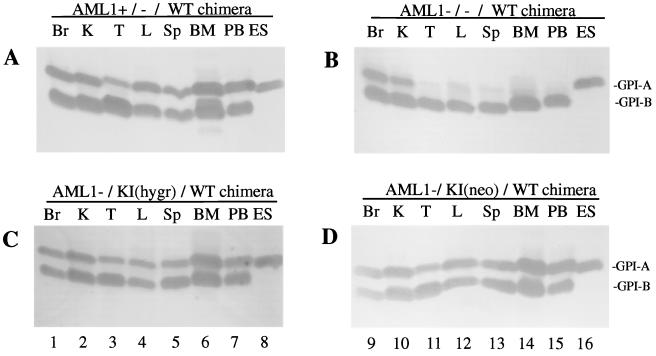
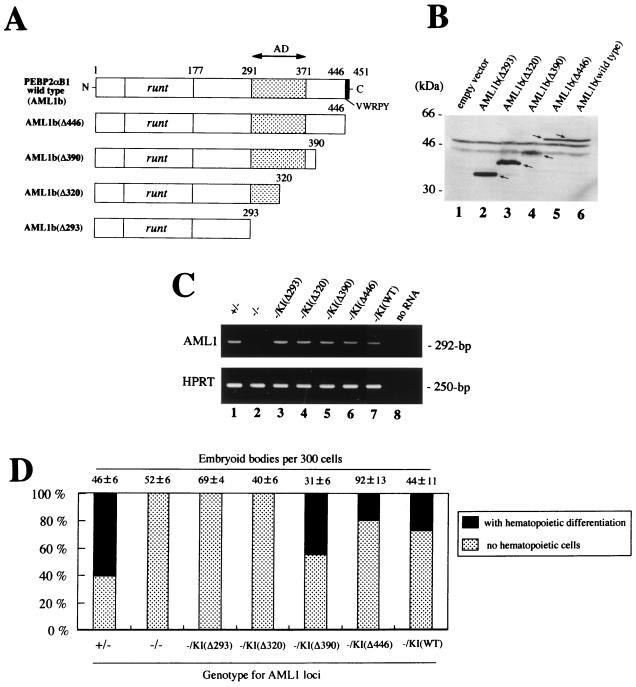
AML1 is one of the most frequently mutated genes associated with human acute leukemia and encodes the DNA-binding subunit of the heterodimering transcriptional factor complex, core-binding factor (CBF) (or polyoma enhancer binding protein 2 [PEBP2]). A null mutation in either AML1 or its dimerizing partner, CBFbeta, results in embryonic lethality secondary to a complete block in fetal liver hematopoiesis, indicating an essential role of this transcription complex in the development of definitive hematopoiesis. The hematopoietic phenotype that results from the loss of AML1 can be replicated in vitro with a two-step culture system of murine embryonic stem (ES) cells. Using this experimental system, we now demonstrate that this hematopoietic defect can be rescued by expressing the PEBP2alphaB1 (AML1b) isoform under the endogenous AML1-regulatory sequences through a knock-in (targeted insertion) approach. Moreover, we demonstrate that the rescued AML1(-/-) ES cell clones contribute to lymphohematopoiesis within the context of chimeric animals. Rescue requires the transcription activation domain of AML1 but does not require the C-terminal VWRPY motif, which is conserved in all AML1 family members and has been shown to interact with the transcriptional corepressor, Groucho/transducin-like Enhancer of split. Taken together, these data provide compelling evidence that the phenotype seen in AML1-deficient mice is due solely to the loss of transcriptionally active AML1.
Figures
Similar articles
-
VWRPY motif-dependent and -independent roles of AML1/Runx1 transcription factor in murine hematopoietic development.Blood. 2004 Jan 15;103(2):562-70. doi: 10.1182/blood-2003-06-2109. Epub 2003 Sep 22. Blood. 2004. PMID: 14504086
-
The transcriptionally active form of AML1 is required for hematopoietic rescue of the AML1-deficient embryonic para-aortic splanchnopleural (P-Sp) region.Blood. 2004 Dec 1;104(12):3558-64. doi: 10.1182/blood-2004-04-1535. Epub 2004 Jul 22. Blood. 2004. PMID: 15271791
-
Identification of an alternatively spliced form of the mouse AML1/RUNX1 gene transcript AML1c and its expression in early hematopoietic development.Biochem Biophys Res Commun. 2001 Mar;281(5):1248-55. doi: 10.1006/bbrc.2001.4513. Biochem Biophys Res Commun. 2001. PMID: 11243869
-
The AML1 gene: a transcription factor involved in the pathogenesis of myeloid and lymphoid leukemias.Haematologica. 1997 May-Jun;82(3):364-70. Haematologica. 1997. PMID: 9234595 Review.
-
The role of the AML1 transcription factor in leukemogenesis.Int J Hematol. 2001 Oct;74(3):258-65. doi: 10.1007/BF02982058. Int J Hematol. 2001. PMID: 11721960 Review.
Cited by
-
Long and short non-coding RNAs as regulators of hematopoietic differentiation.Int J Mol Sci. 2013 Jul 15;14(7):14744-70. doi: 10.3390/ijms140714744. Int J Mol Sci. 2013. PMID: 23860209 Free PMC article. Review.
-
RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making.Crit Rev Oncog. 2011;16(1-2):77-91. doi: 10.1615/critrevoncog.v16.i1-2.80. Crit Rev Oncog. 2011. PMID: 22150309 Free PMC article. Review.
-
Loss of RUNX1/AML1 arginine-methylation impairs peripheral T cell homeostasis.Br J Haematol. 2015 Sep;170(6):859-73. doi: 10.1111/bjh.13499. Epub 2015 May 26. Br J Haematol. 2015. PMID: 26010396 Free PMC article.
-
TGF-β superfamily gene expression and induction of the Runx1 transcription factor in adult neurogenic regions after brain injury.PLoS One. 2013;8(3):e59250. doi: 10.1371/journal.pone.0059250. Epub 2013 Mar 21. PLoS One. 2013. PMID: 23555640 Free PMC article.
-
Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development.Proc Natl Acad Sci U S A. 2001 Jul 17;98(15):8650-5. doi: 10.1073/pnas.151236498. Epub 2001 Jul 3. Proc Natl Acad Sci U S A. 2001. PMID: 11438701 Free PMC article.
References
-
- Bae S C, Yamaguchi-Iwai Y, Ogawa E, Maruyama M, Inuzuka M, Kagoshima H, Shigesada K, Satake M, Ito Y. Isolation of PEBP2 alpha B cDNA representing the mouse homolog of human acute myeloid leukemia gene, AML1. Oncogene. 1993;8:809–814. - PubMed
-
- Daga A, Tighe J E, Calabi F. Leukaemia/Drosophila homology. Nature. 1992;356:484. - PubMed
-
- Downing J R. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol. 1999;106:296–308. - PubMed
-
- Dzierzak E, Medvinsky A. Mouse embryonic hematopoiesis. Trends Genet. 1995;11:359–366. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical