

Estradiol repression of tumor necrosis factor-alpha transcription requires estrogen receptor activation function-2 and is enhanced by coactivators
- PMID: 10611355
- PMCID: PMC24790
- DOI: 10.1073/pnas.96.26.15161
Estradiol repression of tumor necrosis factor-alpha transcription requires estrogen receptor activation function-2 and is enhanced by coactivators
Abstract
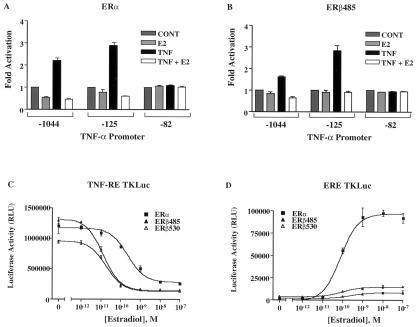
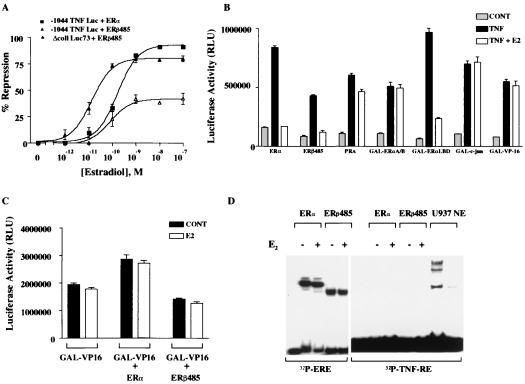
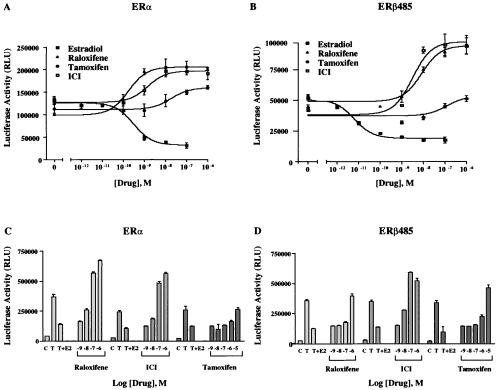
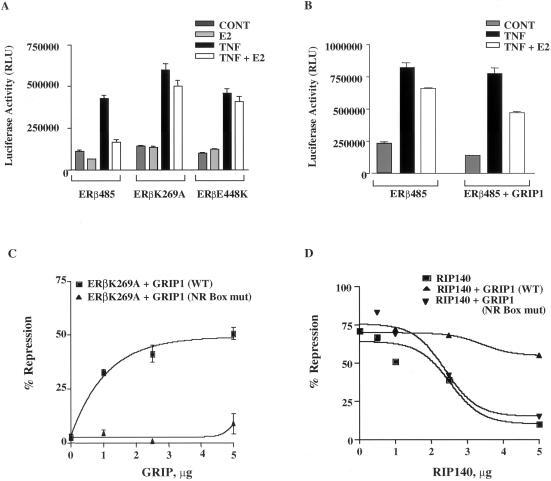
The tumor necrosis factor-alpha (TNF-alpha) promoter was used to explore the molecular mechanisms of estradiol (E(2))-dependent repression of gene transcription. E(2) inhibited basal activity and abolished TNF-alpha activation of the TNF-alpha promoter. The E(2)-inhibitory element was mapped to the -125 to -82 region of the TNF-alpha promoter, known as the TNF-responsive element (TNF-RE). An AP-1-like site in the TNF-RE is essential for repression activity. Estrogen receptor (ER) beta is more potent than ERalpha at repressing the -1044 TNF-alpha promoter and the TNF-RE upstream of the herpes simplex virus thymidine kinase promoter, but weaker at activating transcription through an estrogen response element. The activation function-2 (AF-2) surface in the ligand-binding domain is required for repression, because anti-estrogens and AF-2 mutations impair repression. The requirement of the AF-2 surface for repression is probably due to its capacity to recruit p160 coactivators or related coregulators, because overexpressing the coactivator glucocorticoid receptor interacting protein-1 enhances repression, whereas a glucocorticoid receptor interacting protein-1 mutant unable to interact with the AF-2 surface is ineffective. Furthermore, receptor interacting protein 140 prevents repression by ERbeta, probably by interacting with the AF-2 surface and blocking the binding of endogenous coactivators. These studies demonstrate that E(2)-mediated repression requires the AF-2 surface and the participation of coactivators or other coregulatory proteins.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
