

Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit
- PMID: 10611370
- PMCID: PMC24805
- DOI: 10.1073/pnas.96.26.15245
Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit
Abstract
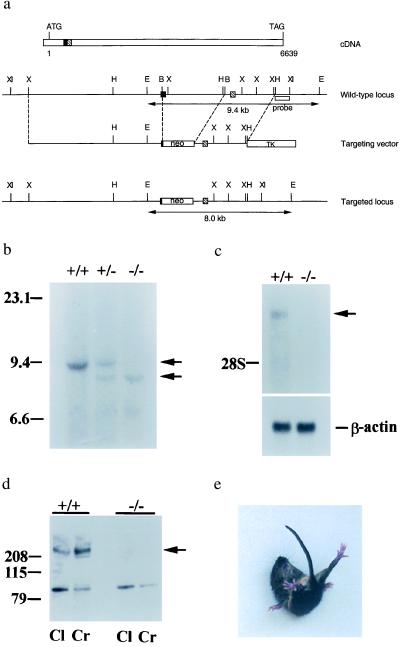
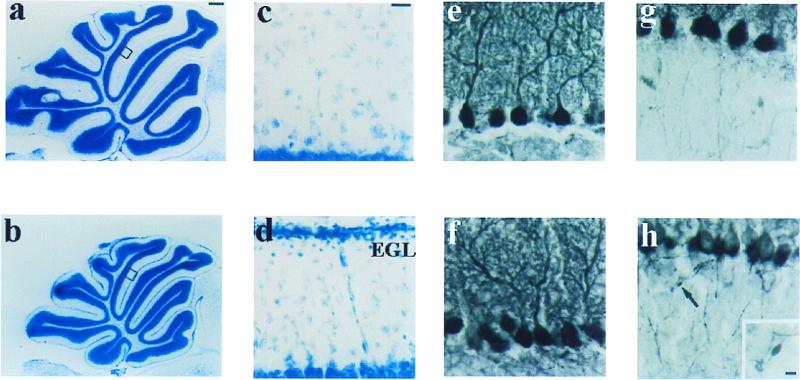
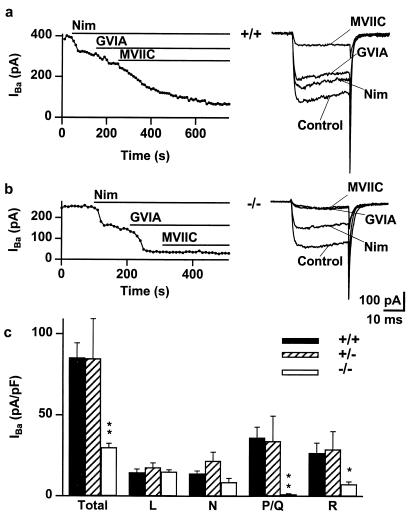
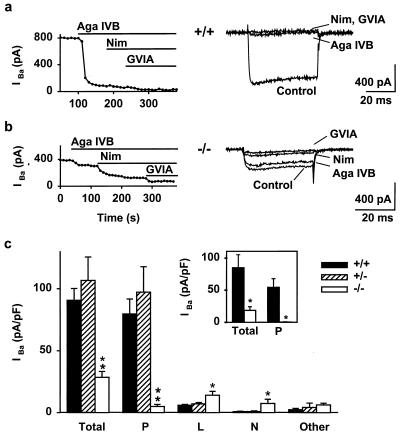
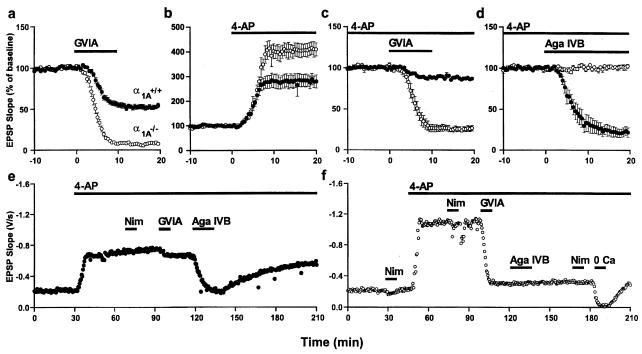
The Ca(2+) channel alpha(1A)-subunit is a voltage-gated, pore-forming membrane protein positioned at the intersection of two important lines of research: one exploring the diversity of Ca(2+) channels and their physiological roles, and the other pursuing mechanisms of ataxia, dystonia, epilepsy, and migraine. alpha(1A)-Subunits are thought to support both P- and Q-type Ca(2+) channel currents, but the most direct test, a null mutant, has not been described, nor is it known which changes in neurotransmission might arise from elimination of the predominant Ca(2+) delivery system at excitatory nerve terminals. We generated alpha(1A)-deficient mice (alpha(1A)(-/-)) and found that they developed a rapidly progressive neurological deficit with specific characteristics of ataxia and dystonia before dying approximately 3-4 weeks after birth. P-type currents in Purkinje neurons and P- and Q-type currents in cerebellar granule cells were eliminated completely whereas other Ca(2+) channel types, including those involved in triggering transmitter release, also underwent concomitant changes in density. Synaptic transmission in alpha(1A)(-/-) hippocampal slices persisted despite the lack of P/Q-type channels but showed enhanced reliance on N-type and R-type Ca(2+) entry. The alpha(1A)(-/-) mice provide a starting point for unraveling neuropathological mechanisms of human diseases generated by mutations in alpha(1A).
Figures
Similar articles
-
Deletion of Cav2.1(alpha1(A)) subunit of Ca2+-channels impairs synaptic GABA and glutamate release in the mouse cerebellar cortex in cultured slices.Eur J Neurosci. 2009 Dec;30(12):2293-307. doi: 10.1111/j.1460-9568.2009.07023.x. Epub 2009 Dec 10. Eur J Neurosci. 2009. PMID: 20092572
-
Impact of the leaner P/Q-type Ca2+ channel mutation on excitatory synaptic transmission in cerebellar Purkinje cells.J Physiol. 2008 Sep 15;586(18):4501-15. doi: 10.1113/jphysiol.2008.156232. Epub 2008 Jul 31. J Physiol. 2008. PMID: 18669535 Free PMC article.
-
Increased expression of P/Q-type Ca2+ channel alpha1A subunit mRNA in cerebellum of N-type Ca2+ channel alpha1B subunit gene-deficient mice.Brain Res Mol Brain Res. 2004 Apr 29;124(1):79-87. doi: 10.1016/j.molbrainres.2004.02.007. Brain Res Mol Brain Res. 2004. PMID: 15093688
-
[P/Q-type voltage-dependent calcium channels in neurological disease].Neurologia. 2007 Oct;22(8):511-6. Neurologia. 2007. PMID: 17573560 Review. Spanish.
-
Calcium channels and channelopathies of the central nervous system.Mol Neurobiol. 2002 Feb;25(1):31-50. doi: 10.1385/MN:25:1:031. Mol Neurobiol. 2002. PMID: 11890456 Review.
Cited by
-
Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons.J Neurosci. 2002 Aug 1;22(15):6362-71. doi: 10.1523/JNEUROSCI.22-15-06362.2002. J Neurosci. 2002. PMID: 12151514 Free PMC article.
-
Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response.J Neurosci. 2004 Jun 9;24(23):5400-9. doi: 10.1523/JNEUROSCI.0553-04.2004. J Neurosci. 2004. PMID: 15190113 Free PMC article.
-
Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel.EMBO J. 2001 May 15;20(10):2349-56. doi: 10.1093/emboj/20.10.2349. EMBO J. 2001. PMID: 11350923 Free PMC article.
-
Pedunculopontine arousal system physiology - Implications for insomnia.Sleep Sci. 2015 Apr-Jun;8(2):92-9. doi: 10.1016/j.slsci.2015.06.002. Epub 2015 Jul 10. Sleep Sci. 2015. PMID: 26483950 Free PMC article. Review.
-
Ca(2+) current facilitation determines short-term facilitation at inhibitory synapses between cerebellar Purkinje cells.J Physiol. 2015 Nov 15;593(22):4889-904. doi: 10.1113/JP270704. Epub 2015 Oct 12. J Physiol. 2015. PMID: 26337248 Free PMC article.
References
-
- Mori Y, Friedrich T, Kim M S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, et al. Nature (London) 1991;350:398–402. - PubMed
-
- Dunlap K, Luebke J I, Turner T J. Trends Neurosci. 1995;18:89–98. - PubMed
-
- De Waard M, Gurnett C A, Campbell K P. In: Ion Channels. Narahashi T, editor. Vol. 4. New York: Plenum; 1996. pp. 41–87. - PubMed
-
- Catterall W A. Cell Calcium. 1998;24:307–23. - PubMed
-
- Wheeler D B, Tsien R W. In: Calcium as a Cellular Regulator. Carafoli E, Lee C, editors. New York: Oxford Univ. Press; 1999. pp. 171–199.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
