

Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase
- PMID: 10613903
- PMCID: PMC2174254
- DOI: 10.1083/jcb.147.7.1443
Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase
Abstract
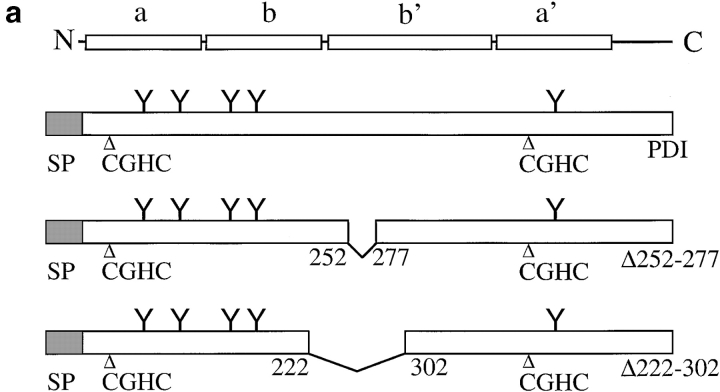
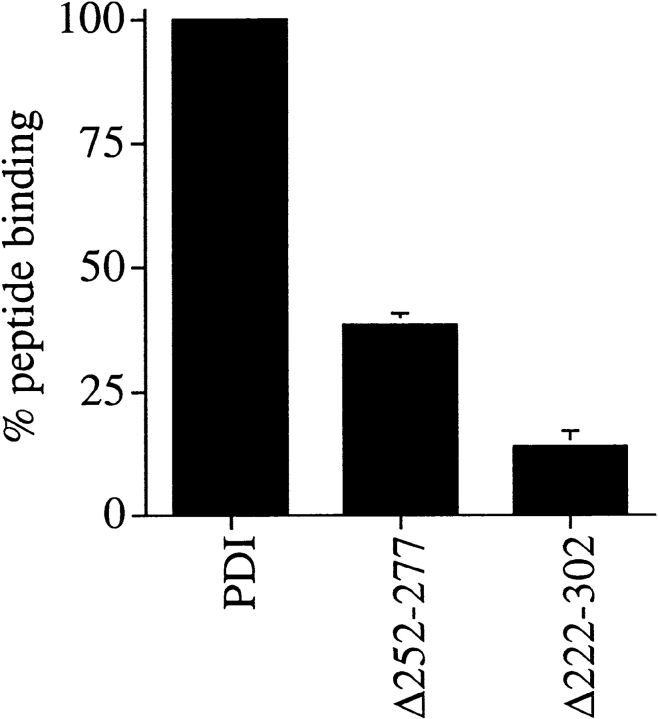
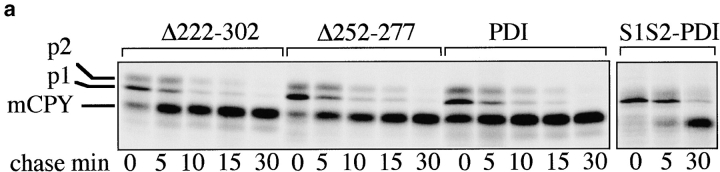
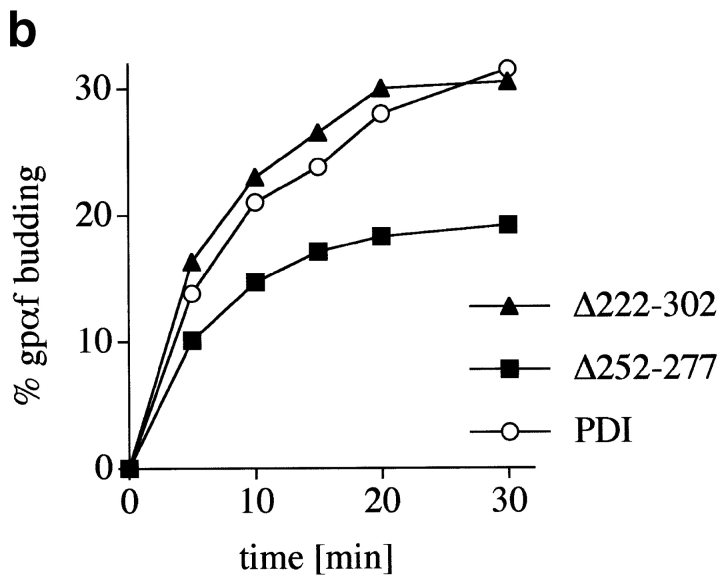
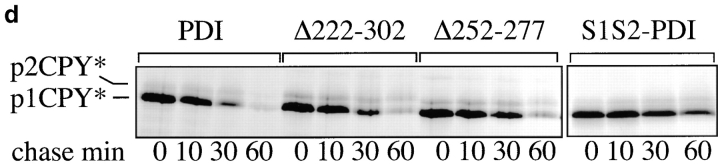
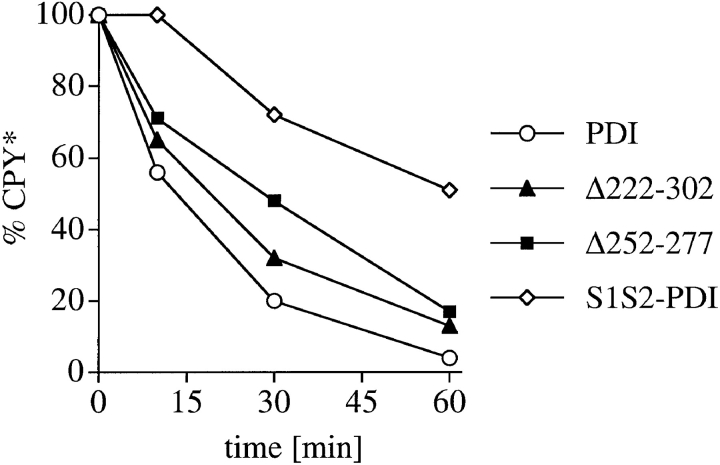
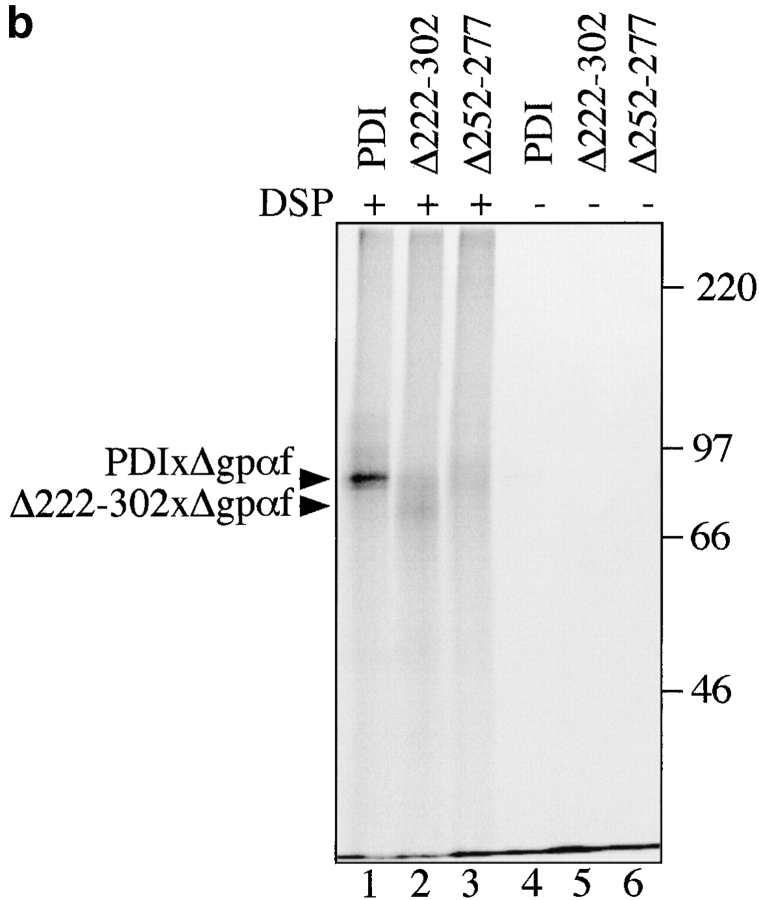
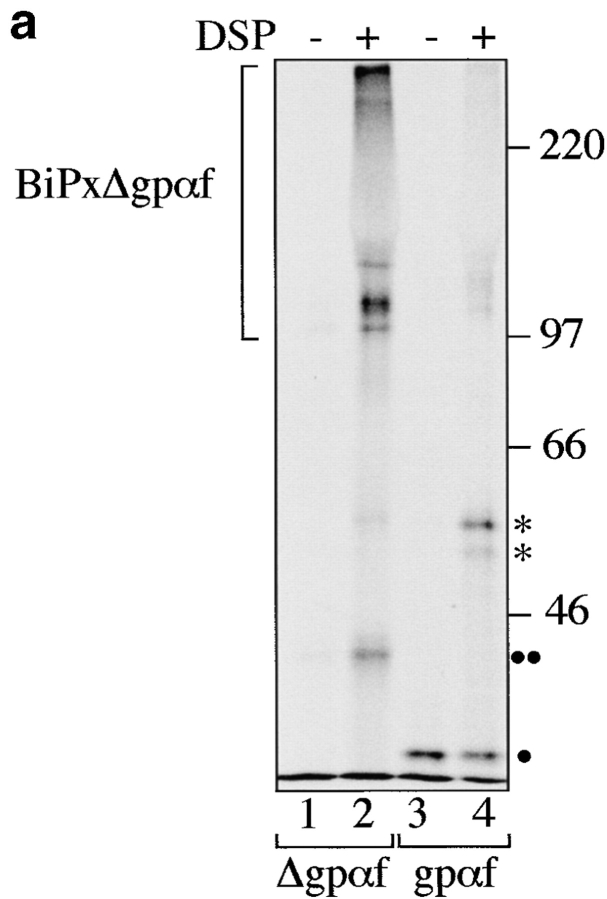
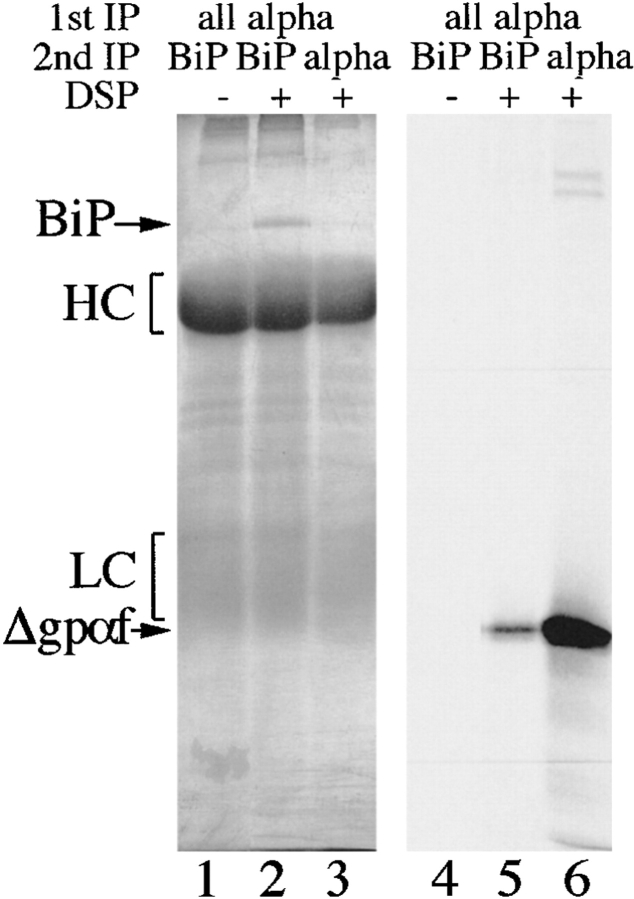
Protein disulfide isomerase (PDI) interacts with secretory proteins, irrespective of their thiol content, late during translocation into the ER; thus, PDI may be part of the quality control machinery in the ER. We used yeast pdi1 mutants with deletions in the putative peptide binding region of the molecule to investigate its role in the recognition of misfolded secretory proteins in the ER and their export to the cytosol for degradation. Our pdi1 deletion mutants are deficient in the export of a misfolded cysteine-free secretory protein across the ER membrane to the cytosol for degradation, but ER-to-Golgi complex transport of properly folded secretory proteins is only marginally affected. We demonstrate by chemical cross-linking that PDI specifically interacts with the misfolded secretory protein and that mutant forms of PDI have a lower affinity for this protein. In the ER of the pdi1 mutants, a higher proportion of the misfolded secretory protein remains associated with BiP, and in export-deficient sec61 mutants, the misfolded secretory protein remain bounds to PDI. We conclude that the chaperone PDI is part of the quality control machinery in the ER that recognizes terminally misfolded secretory proteins and targets them to the export channel in the ER membrane.
Figures












References
-
- Andrews D.W., Johnson A.E. The transloconmore than a hole in the membrane? Trends Biochem. Sci. 1996;21:365–369. - PubMed
-
- Baker D., Hicke L., Rexach M., Schleyer M., Schekman R. Reconstitution of SEC gene product dependent intercompartmental protein transport. Cell. 1988;54:335–344. - PubMed
-
- Brodsky J.L., Werner E.D., Dubas M.E., Goeckeler J.L., Kruse K.B., McCracken A.A. The requirement for molecular chaperones during endoplasmic reticulum associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 1999;274:3453–3460. - PubMed
-
- Bulleid N.J., Freedman R.B. Defective cotranslational formation of disulphide-bonds in protein disulphide-isomerase-deficient microsomes. Nature. 1988;335:649–651. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
