

Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists
- PMID: 10632585
- PMCID: PMC6772401
- DOI: 10.1523/JNEUROSCI.20-02-00558.2000
Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists
Abstract
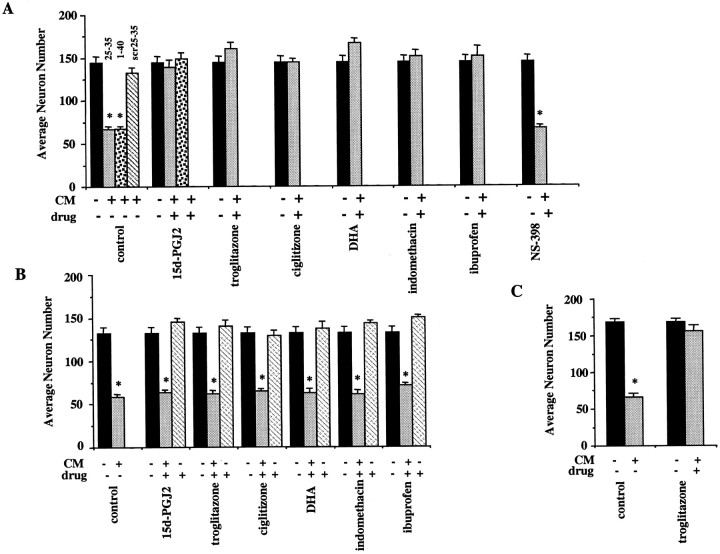
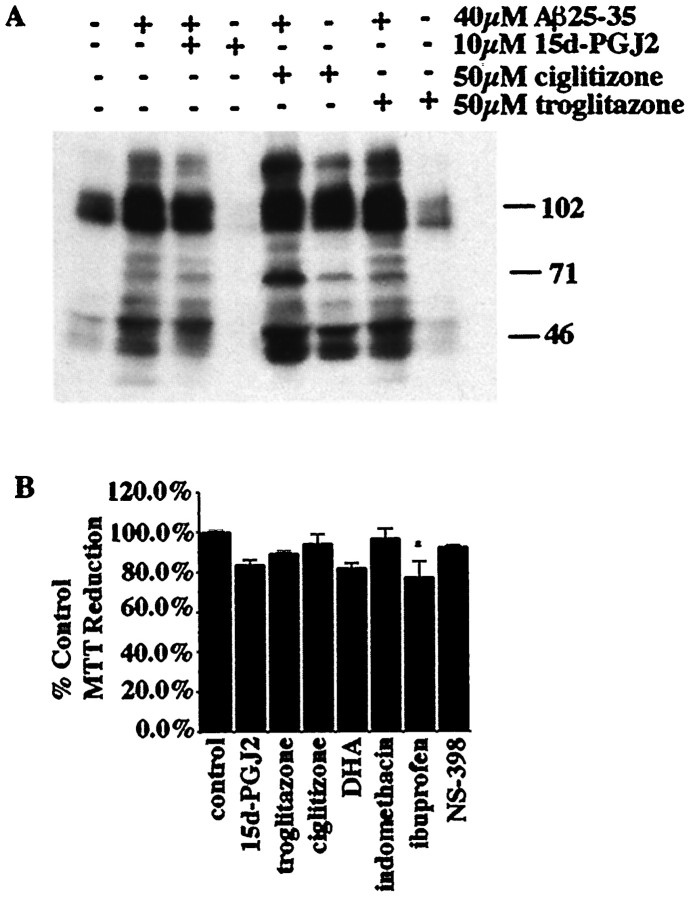
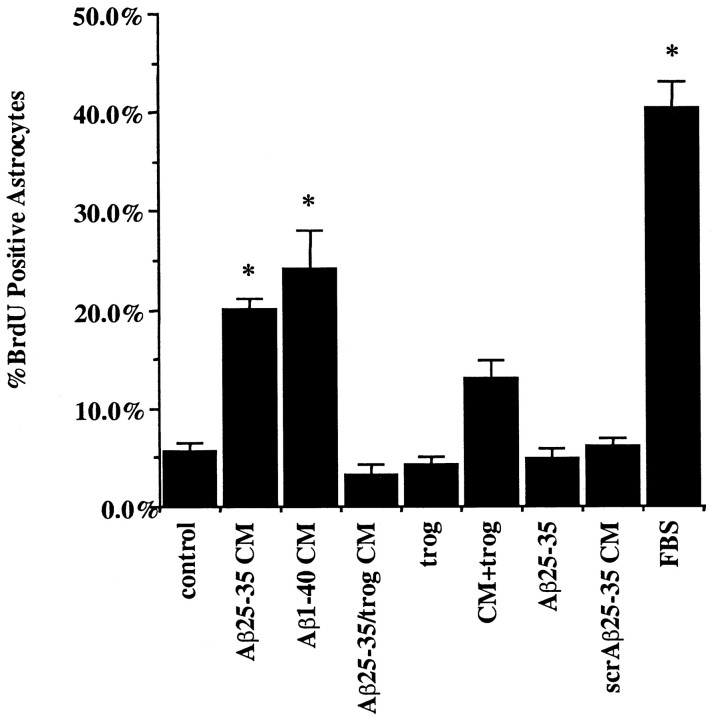
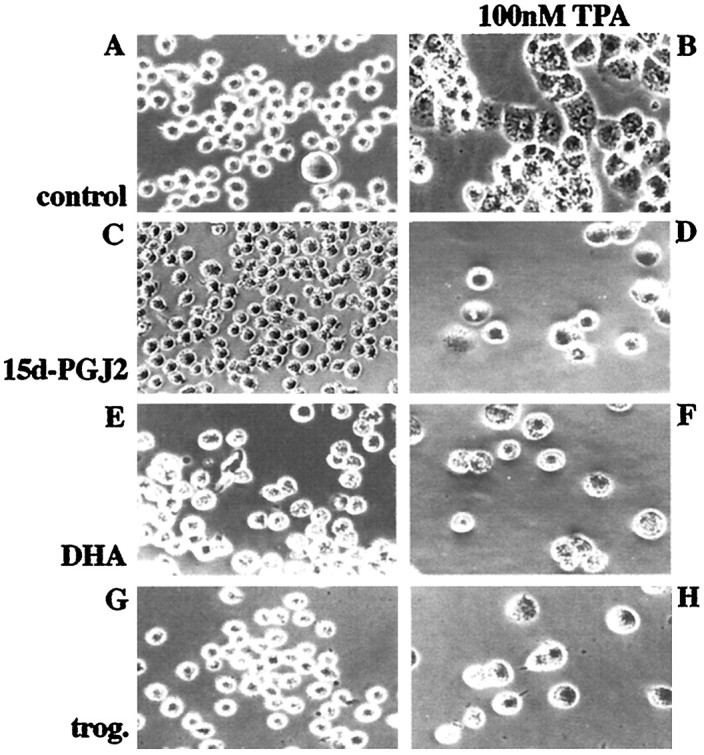
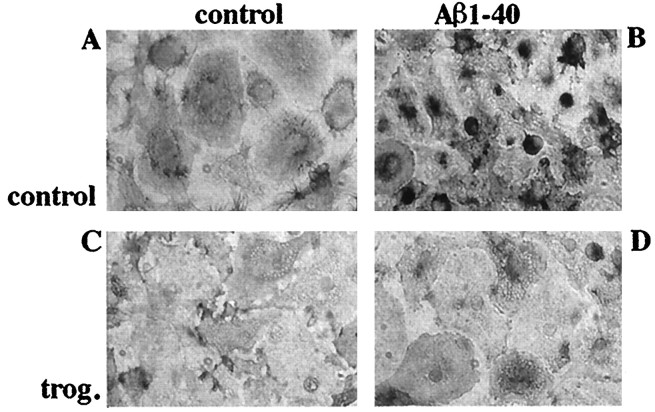
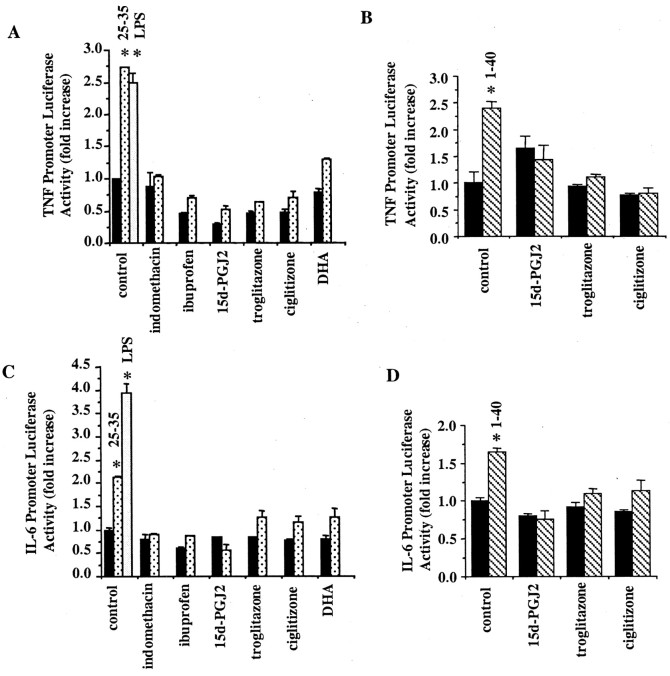
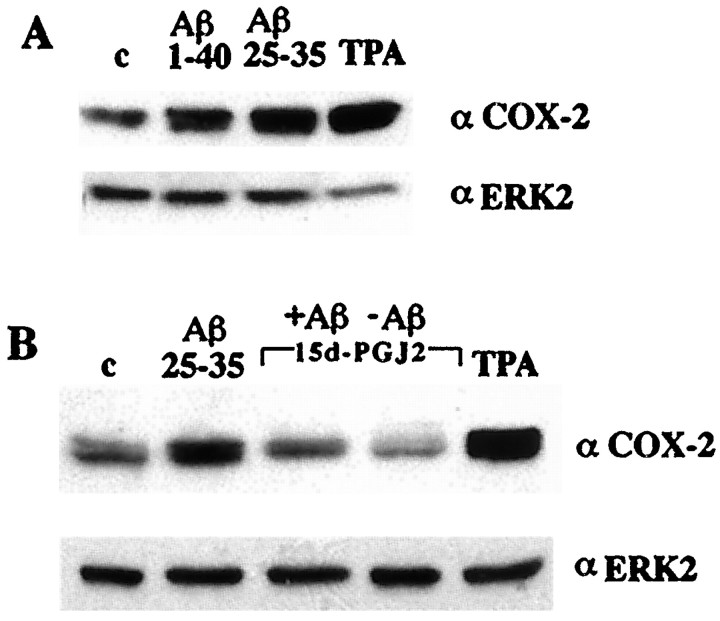
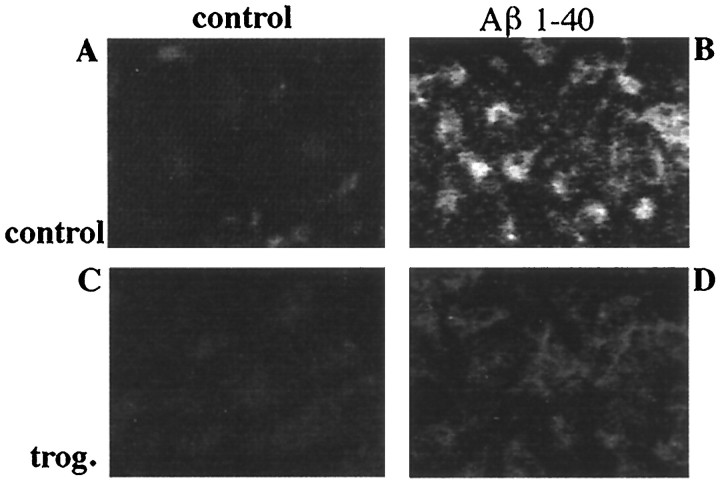
Alzheimer's disease (AD) is characterized by the extracellular deposition of beta-amyloid fibrils within the brain and the subsequent association and phenotypic activation of microglial cells associated with the amyloid plaque. The activated microglia mount a complex local proinflammatory response with the secretion of a diverse range of inflammatory products. Nonsteroidal anti-inflammatory drugs (NSAIDs) are efficacious in reducing the incidence and risk of AD and significantly delaying disease progression. A recently appreciated target of NSAIDs is the ligand-activated nuclear receptor peroxisome proliferator-activated receptor gamma (PPARgamma). PPARgamma is a DNA-binding transcription factor whose transcriptional regulatory actions are activated after agonist binding. We report that NSAIDs, drugs of the thiazolidinedione class, and the natural ligand prostaglandin J2 act as agonists for PPARgamma and inhibit the beta-amyloid-stimulated secretion of proinflammatory products by microglia and monocytes responsible for neurotoxicity and astrocyte activation. The activation of PPARgamma also arrested the differentiation of monocytes into activated macrophages. PPARgamma agonists were shown to inhibit the beta-amyloid-stimulated expression of the cytokine genes interleukin-6 and tumor necrosis factor alpha. Furthermore, PPARgamma agonists inhibited the expression of cyclooxygenase-2. These data provide direct evidence that PPARgamma plays a critical role in regulating the inflammatory responses of microglia and monocytes to beta-amyloid. We argue that the efficacy of NSAIDs in the treatment of AD may be a consequence of their actions on PPARgamma rather than on their canonical targets the cyclooxygenases. Importantly, the efficacy of these agents in inhibiting a broad range of inflammatory responses suggests PPARgamma agonists may provide a novel therapeutic approach to AD.
Figures
References
-
- Aisen PS. Inflammation and Alzheimer's disease: mechanisms and therapeutic strategies. Gerontology. 1997;43:143–149. - PubMed
-
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia. 1993;7:111–118. - PubMed
-
- Berg L, McKeel DW, Jr, Miller JP, Baty J, Morris JC. Neuropathological indexes of Alzheimer's disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–358. - PubMed
-
- Bjorkman DJ. The effect of aspirin and nonsteroidal anti-inflammatory drugs on prostaglandins. Am J Med. 1998;105:8S–12S. - PubMed
-
- Bolten WW. Scientific rationale for specific inhibition of COX-2. J Rheumatol Suppl. 1998;51:2–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials