

Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha
- PMID: 10640274
- PMCID: PMC316350
Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha
Abstract
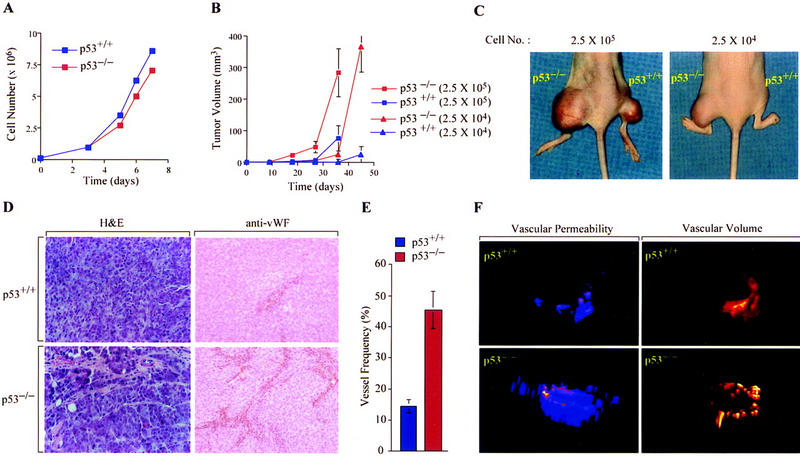
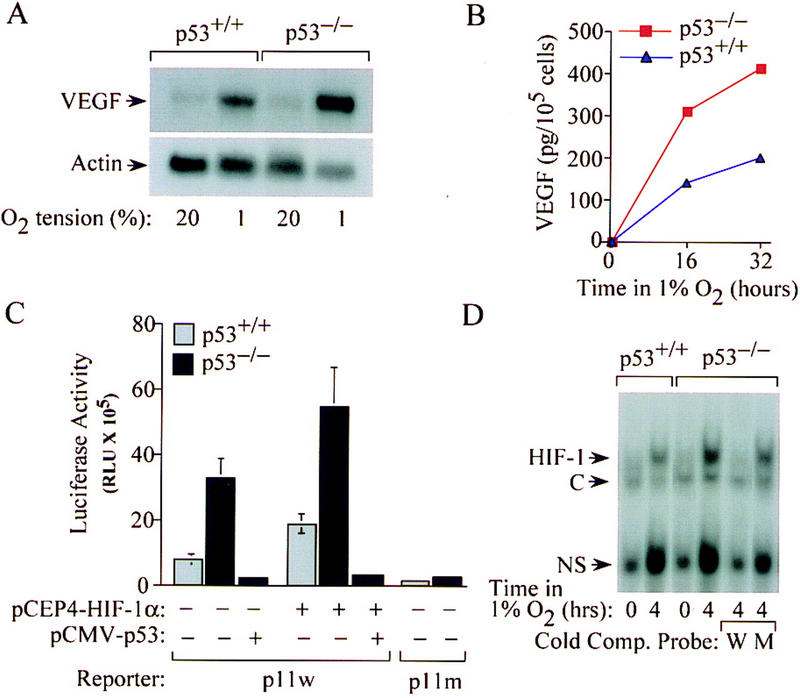
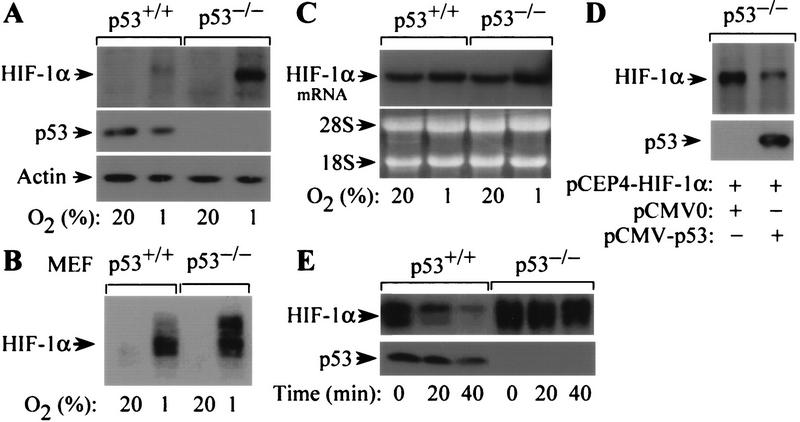
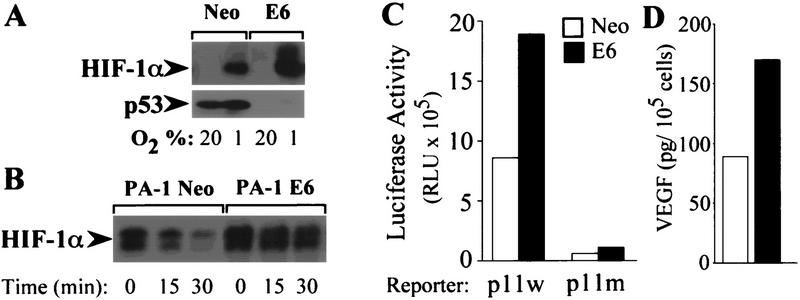
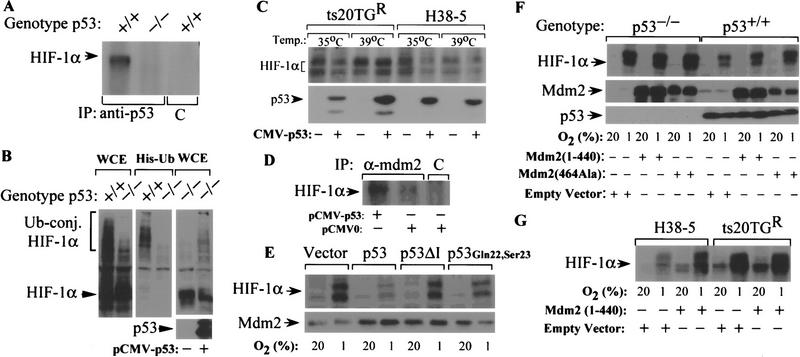
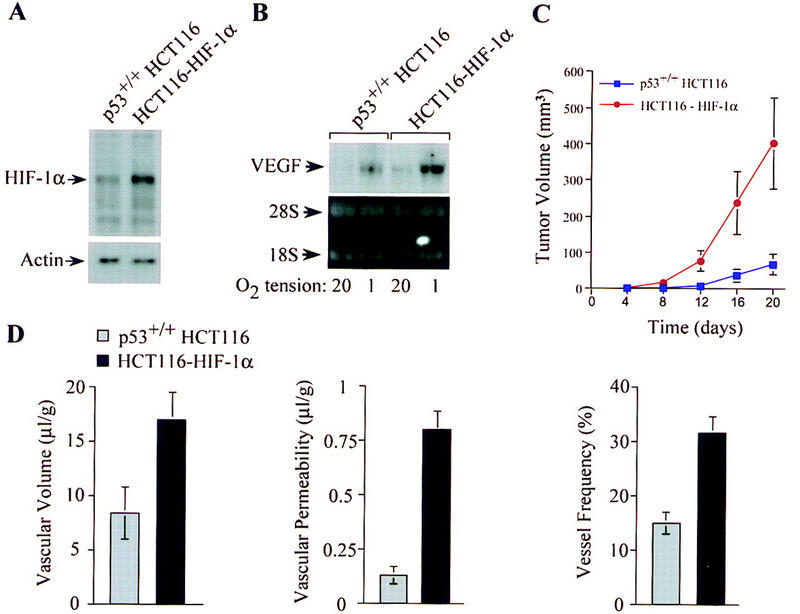
The switch to an angiogenic phenotype is a fundamental determinant of neoplastic growth and tumor progression. We demonstrate that homozygous deletion of the p53 tumor suppressor gene via homologous recombination in a human cancer cell line promotes the neovascularization and growth of tumor xenografts in nude mice. We find that p53 promotes Mdm2-mediated ubiquitination and proteasomal degradation of the HIF-1alpha subunit of hypoxia-inducible factor 1 (HIF-1), a heterodimeric transcription factor that regulates cellular energy metabolism and angiogenesis in response to oxygen deprivation. Loss of p53 in tumor cells enhances HIF-1alpha levels and augments HIF-1-dependent transcriptional activation of the vascular endothelial growth factor (VEGF) gene in response to hypoxia. Forced expression of HIF-1alpha in p53-expressing tumor cells increases hypoxia-induced VEGF expression and augments neovascularization and growth of tumor xenografts. These results indicate that amplification of normal HIF-1-dependent responses to hypoxia via loss of p53 function contributes to the angiogenic switch during tumorigenesis.
Figures
References
-
- An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature. 1998;392:405–408. - PubMed
-
- Bergers G, Javaherian K, Lo KN, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999;284:808–812. - PubMed
-
- Blagosklonny MV, An WG, Romanova LY, Trepel J, Fojo T, Neckers L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J Biol Chem. 1998;273:11995–11998. - PubMed
-
- Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous