

Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie)
- PMID: 10642597
- PMCID: PMC377427
- DOI: 10.1172/JCI7302
Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie)
Abstract
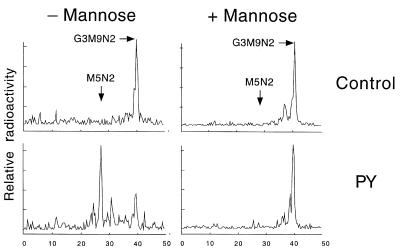
Congenital disorders of glycosylation (CDGs) are metabolic deficiencies in glycoprotein biosynthesis that usually cause severe mental and psychomotor retardation. Different forms of CDGs can be recognized by altered isoelectric focusing (IEF) patterns of serum transferrin (Tf). Two patients with these symptoms and similar abnormal Tf IEF patterns were analyzed by metabolic labeling of fibroblasts with ¿2-(3)Hmannose. The patients produced a truncated dolichol-linked precursor oligosaccharide with 5 mannose residues, instead of the normal precursor with 9 mannose residues. Addition of 250 microM mannose to the culture medium corrected the size of the truncated oligosaccharide. Microsomes from fibroblasts of these patients were approximately 95% deficient in dolichol-phosphate-mannose (Dol-P-Man) synthase activity, with an apparent K(m) for GDP-Man approximately 6-fold higher than normal. DPM1, the gene coding for the catalytic subunit of Dol-P-Man synthase, was altered in both patients. One patient had a point mutation, C(274)G, causing an R(92)G change in the coding sequence. The other patient also had the C(274)G mutation and a 13-bp deletion that presumably resulted in an unstable transcript. Defects in DPM1 define a new glycosylation disorder, CDG-Ie.
Figures




Comment in
-
Congenital disorders of glycosylation caused by defects in mannose addition during N-linked oligosaccharide assembly.J Clin Invest. 2000 Jan;105(2):131-2. doi: 10.1172/JCI9157. J Clin Invest. 2000. PMID: 10642590 Free PMC article. No abstract available.
References
-
- Freeze HH. Disorders in protein glycosylation and potential therapy: tip of an iceberg? J Pediatr. 1998;133:593–600. - PubMed
-
- Krasnewich D, Gahl WA. Carbohydrate-deficient glycoprotein syndrome. Adv Pediatr. 1997;44:109–140. - PubMed
-
- Jaeken J, Stibler H, Hagberg B. The carbohydrate-deficient glycoprotein syndrome. A new inherited multisystemic disease with severe nervous system involvement. Acta Paediatr Scand Suppl. 1991;375:1–71. - PubMed
-
- Matthijs G, et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome. Nat Genet. 1997;16:88–92. - PubMed
-
- Van Schaftingen E, Jaeken J. Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett. 1995;377:318–320. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
