

Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie
- PMID: 10642602
- PMCID: PMC377434
- DOI: 10.1172/JCI8691
Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie
Abstract
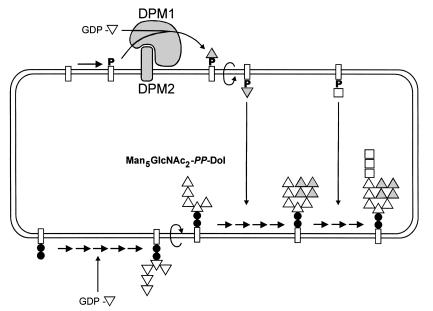
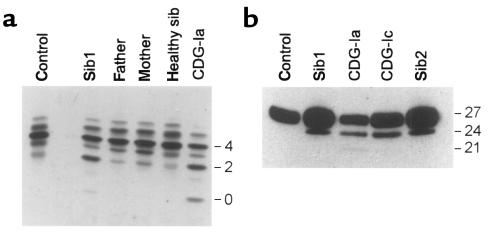
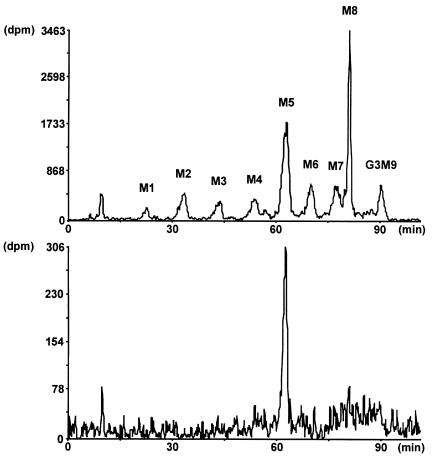
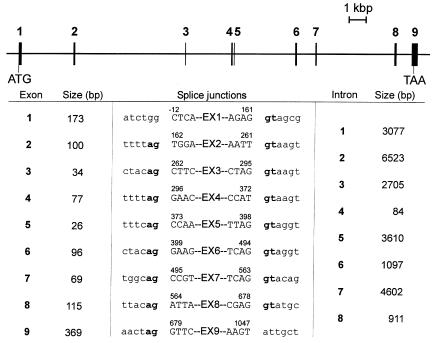
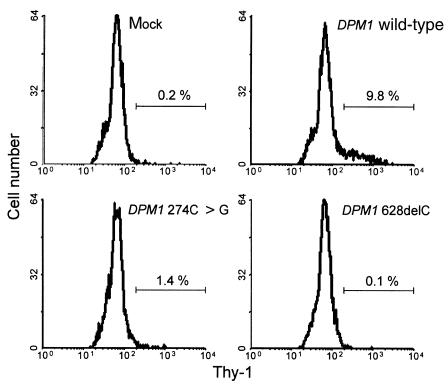
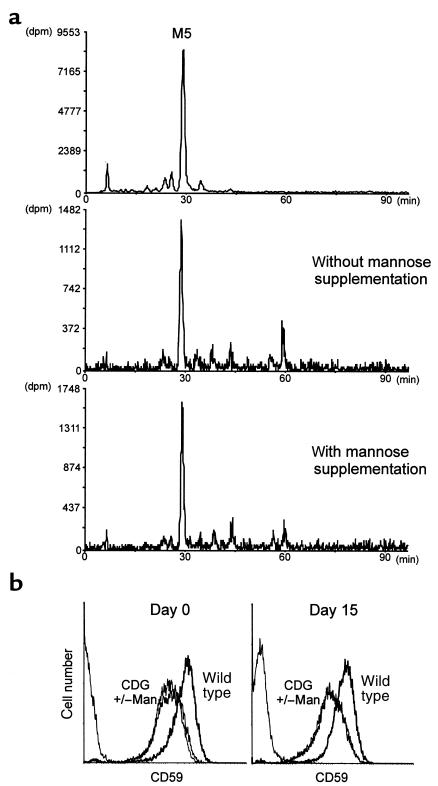
Congenital disorders of glycosylation (CDG), formerly known as carbohydrate-deficient glycoprotein syndromes, lead to diseases with variable clinical pictures. We report the delineation of a novel type of CDG identified in 2 children presenting with severe developmental delay, seizures, and dysmorphic features. We detected hypoglycosylation on serum transferrin and cerebrospinal fluid beta-trace protein. Lipid-linked oligosaccharides in the endoplasmic reticulum of patient fibroblasts showed an accumulation of the dolichyl pyrophosphate Man(5)GlcNAc(2) structure, compatible with the reduced dolichol-phosphate-mannose synthase (DolP-Man synthase) activity detected in these patients. Accordingly, 2 mutant alleles of the DolP-Man synthase DPM1 gene, 1 with a 274C>G transversion, the other with a 628delC deletion, were detected in both siblings. Complementation analysis using DPM1-null murine Thy1-deficient cells confirmed the detrimental effect of both mutations on the enzymatic activity. Furthermore, mannose supplementation failed to improve the glycosylation status of DPM1-deficient fibroblast cells, thus precluding a possible therapeutic application of mannose in the patients. Because DPM1 deficiency, like other subtypes of CDG-I, impairs the assembly of N-glycans, this novel glycosylation defect was named CDG-Ie.
Figures

Comment in
-
Congenital disorders of glycosylation caused by defects in mannose addition during N-linked oligosaccharide assembly.J Clin Invest. 2000 Jan;105(2):131-2. doi: 10.1172/JCI9157. J Clin Invest. 2000. PMID: 10642590 Free PMC article. No abstract available.
References
-
- Jaeken J, Carchon H, Stibler H. The carbohydrate-deficient glycoprotein syndromes: pre-Golgi and Golgi disorders? Glycobiology. 1993;3:423–428. - PubMed
-
- Krasnewich D, Gahl WA. Carbohydrate-deficient glycoprotein syndrome. Adv Pediatr. 1997;44:109–140. - PubMed
-
- Matthijs G, et al. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat Genet. 1997;16:88–92. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
