

NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase
- PMID: 10648614
- PMCID: PMC85265
- DOI: 10.1128/MCB.20.4.1278-1290.2000
NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase
Abstract
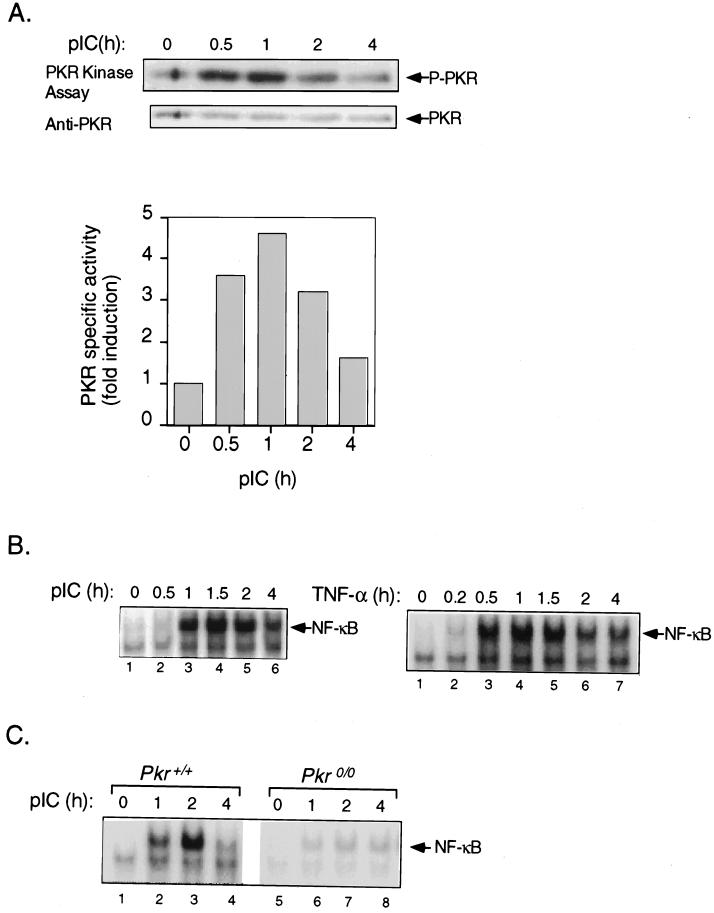
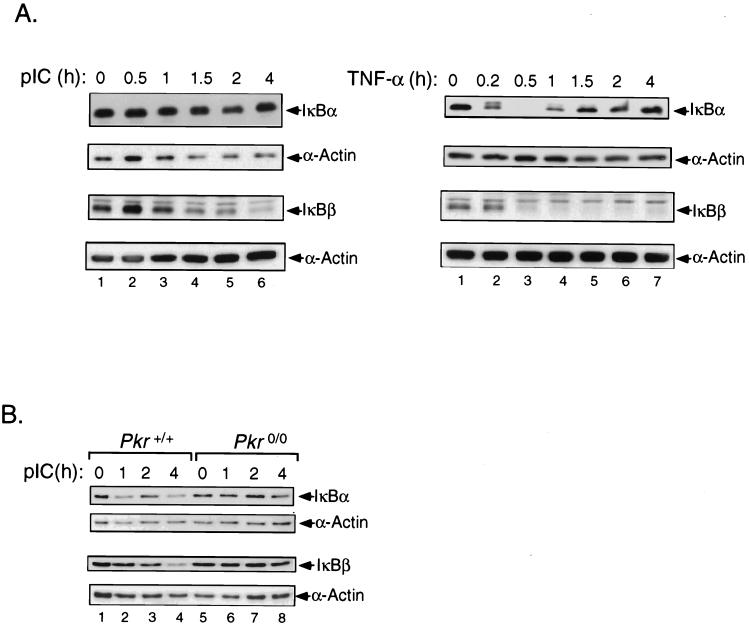
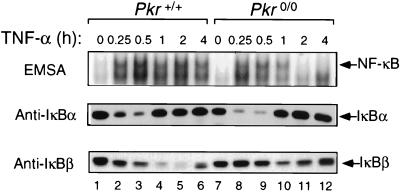
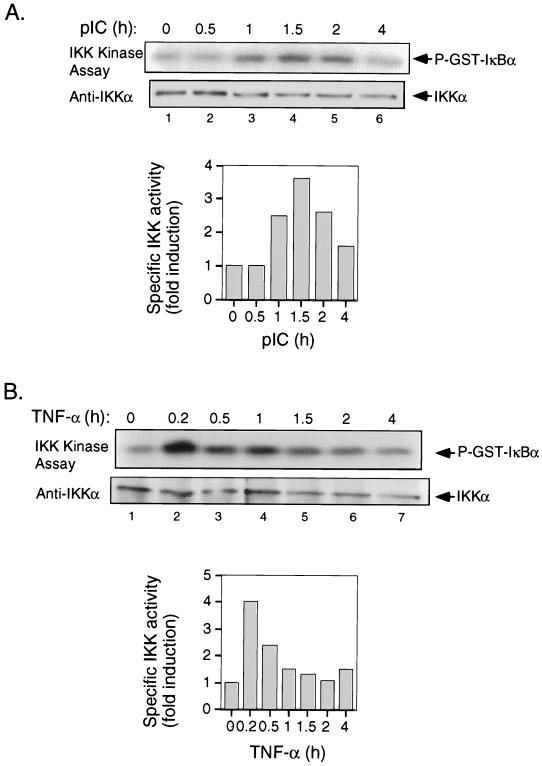
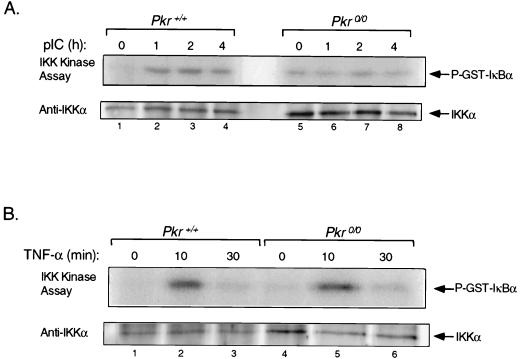
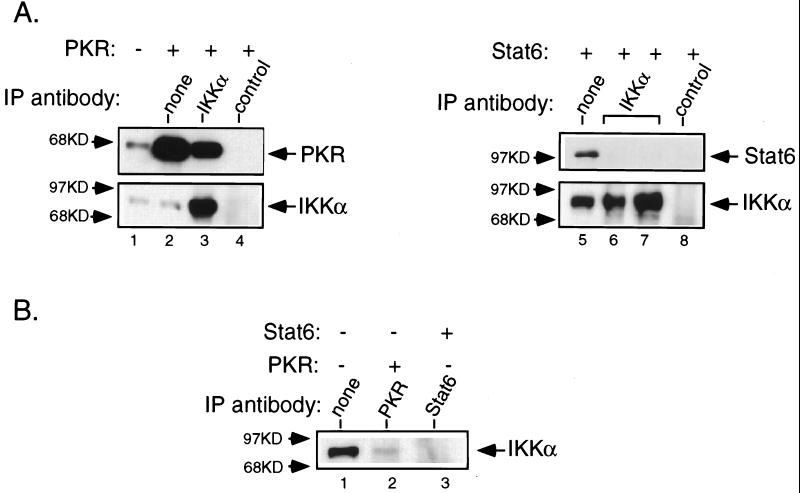
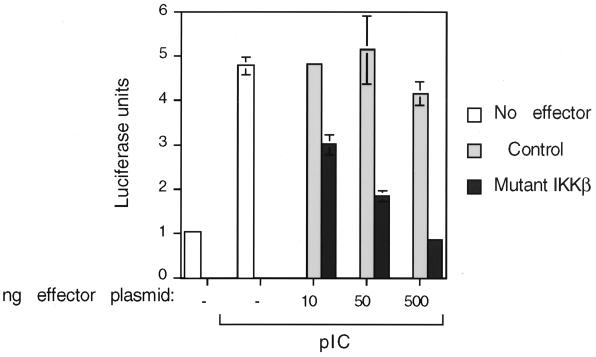
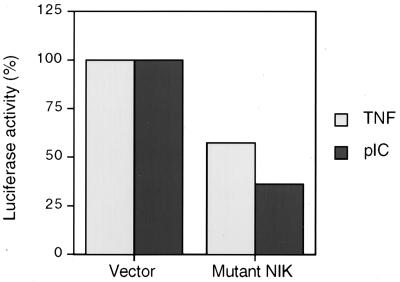
The interferon (IFN)-inducible double-stranded-RNA (dsRNA)-activated serine-threonine protein kinase (PKR) is a major mediator of the antiviral and antiproliferative activities of IFNs. PKR has been implicated in different stress-induced signaling pathways including dsRNA signaling to nuclear factor kappa B (NF-kappaB). The mechanism by which PKR mediates activation of NF-kappaB is unknown. Here we show that in response to poly(rI). poly(rC) (pIC), PKR activates IkappaB kinase (IKK), leading to the degradation of the inhibitors IkappaBalpha and IkappaBbeta and the concomitant release of NF-kappaB. The results of kinetic studies revealed that pIC induced a slow and prolonged activation of IKK, which was preceded by PKR activation. In PKR null cell lines, pIC failed to stimulate IKK activity compared to cells from an isogenic background wild type for PKR in accord with the inability of PKR null cells to induce NF-kappaB in response to pIC. Moreover, PKR was required to establish a sustained response to tumor necrosis factor alpha (TNF-alpha) and to potentiate activation of NF-kappaB by cotreatment with TNF-alpha and IFN-gamma. By coimmunoprecipitation, PKR was shown to be physically associated with the IKK complex. Transient expression of a dominant negative mutant of IKKbeta or the NF-kappaB-inducing kinase (NIK) inhibited pIC-induced gene expression from an NF-kappaB-dependent reporter construct. Taken together, these results demonstrate that PKR-dependent dsRNA induction of NF-kappaB is mediated by NIK and IKK activation.
Figures

References
-
- Baldwin A S., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous